Illumina surprised pretty much everyone with their latest SBS chemistry. I was at an Illumina meeting last week where it was discussed at some length, with many in the audience concerned about possible new error modes due to the drastic change compared to the four-colour SBS used in HiSeq and MiSeq.
I posted an explanation of how the two-colour chemistry works a couple of weeks ago and my initial thoughts remain the same: the chemistry is likely to be an important step forward and Illumina are unlikely to have released it without a lot of confidence in it. So do we need to be particularly concerned about the lack of G signal or the dual-flurophore approach used for A?
Help, my G is missing: argumentum ad ignorantiam, "absence of evidence is not evidence of absence" is the point of view most people seem to have come from. There is concern that a null signal is not the same as a nice bright green spot. But in discussing this with colleagues I was struck by the fact that twenty years ago the lack of signal was the basis for Sanger sequencing working at all. Take a look at the image below, you can read the sequence yourself:
GGTCCGCCACTTCCCGTTAGTC
Anyone who remembers, or actually performed, radioactive sequencing will know what I am talking about. All those lanes to interrogate, reading bases out to a mate while guided by a ruler down the autoradiograph. A genome felt like an impossibility. But the "missing" signal was not an issue, but rather the key to success. 75% of bases in each track had no signal, so what’s all the fuss over the missing G?
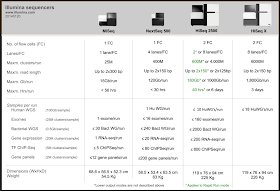
But what about comparisons to HiSeq SBS: We'll have to wait for academic groups to sequence larger sample numbers than Illumina have so far produced to get a real handle on how well the SBS chemistry's compare. For now most data is still going to come from four-colour SBS. But in the future who knows. Will two-colour SBS trickle down/across to other platforms? I'm certainly liking the simplification of hardware and looking forward to a more robust instrument. After all most of my groups Tweets are to say how long our queue is or that our HiSeq has broken.
I only ever did radioactive sequencing once during my third year at Uni, it was a two week long exercise, what with development of the autoradiograph, and I got a few hundred base pairs for my project. That was in 1995, nineteen years later and in two weeks an XTen system could generate 640 30x Human genomes.