Congratulations to Tatiana Borodina who has won the Core Genomics Christmas competition and a free MiSeq run courtesy of Illumina. Special mentions to runners up Tim Forshew (Cambridge Institute) and Charles Warden (City of Hope) who were very close to Tatiana. And also to Thomas Rio Frio (Institut Curie) who was the only person to get all 6 baubles right.
Tatiana graduated from Moscow State University in 2001 and received her PhD from the Max-Planck Institute for Molecular Genetics in 2005 in Prof. H. Lehrach's group. She's been working with NGS from 2007 with Illumina and SOLiD sequencing and since 2012 has been Head of Sequencing at Alacris Theranostics. Tatiana's been reading the Core Genomics blog for about two years and likes it as much for the thoroughness as the way it's written. Thanks for the compliments!
The competition answers: The results make for some interesting reading. 67 people entered the competition from a pretty even mix of wet- and dry-lab users, three-quarters of whom use instruments within their Institute. The post received 1289 views putting it just outside of the top ten posts of 2013.
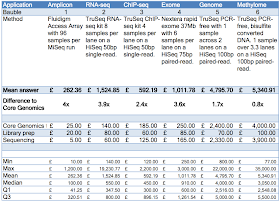
Answers to the competition questions are in the table at the bottom of this post along with methods for how the winner was calculated.
What was what: The baubles were; 1 - Amplicon, 2 - RNA-seq, 3 - ChIP-seq, 4 - Exome, 5 - Genome, 6 - Methylome. The guesses were pretty accurate with 60-75% of people correctly matching the numbers correctly. However ChIP-seq and RNA-seq seem to have caused some difficulties and were mixed up by most entrants. I discuss the main reason I can see in the RNA-seq bullet below.
How much do the different applications cost: I tried to make this as fair as possible by stating the depth of coverage for genomes and methylomes, specifying differential gene expression for RNA-seq and setting the number of amplicons per sample at 48. The pricing I used in setting this competition was that used in my lab; this is the price we pay for a library-prep or sequencing lane in a core facility lab that recovers costs; all consumables, most of the labour but no depreciation or infrastructure. Although everyone might argue my price is not indicative of the true costs I defy anyone to come up with a fair price we can all agree on!
Needless to say the answers to “How much do you think the total cost of sequencing is for each application, i.e. library prep PLUS sequencing?” were hugely variable. I was surprised that only methylomes came out as costing about the same as my estimate (answers averaged out to £2500-£5500). Genomes were almost the same averaging out at 1.7x more than my estimate (answers averaged out to £2500-£5000). But the most surprising thing to me was how entrants thought the other applications were 3-4 fold more expensive than my estimates.
So why do people think NGS costs so much: Either people are paying a lot more for the same data, are getting more/different data than my lab generates, or just guessed and don’t really know. I suspect more/different is behind most cases as the number of samples multiplexed by lane has a huge impact on final costs and this will be determined by read depth and run-type (single vs paired, etc).
Whilst people are likely to debate the true costs of sequencing (Sboner et al), they’ll argue forever over the right sort of sequencing to do! And for RNA-seq and ChIP-seq I suspect this was what caused so many entrants to swap the baubles around.
- Genomes: (£4795, answers averaged out to £2500-£5000) about 1.7x more than my estimate.
- Methylomes: (£5340, answers averaged out to £2500-£5500) pretty close to my estimate but surprisingly you thought a methylome would only be £500 more than a genome.
- Exomes: (£1011, answers averaged out to £600-1200) were massively higher than my lab’s internal costs but the price charged very much depends on the number of samples being processed and the kit being used. Illumina made a massive impact in 2011 when they dropped the price of exomes through the floor, and large pre-capture pooling is making it easier and easier to prepare large number of exome-seq libraries. Hopefully costs are at a point now where replication of exome library prep will become the norm. This should make a big difference to false-positive calls and reduce the false-negative rate a little as well.
- ChIP-seq: (£592, answers averaged out to £300-900) is the oldest and most established method with ENCODE setting guidelines pretty close to what the majority of labs are doing: 10-50M short single-end reads. In my lab we aim for 40M SE50bp reads. However a 5-fold difference in read depth makes a huge difference to the final cost.
- RNA-seq: (£1524, answers averaged out to £350-950) however, is a different matter with a lengthy, and as yet unsettled, debate over single- vs paired-reads; and endless discussion about read-depth (20-200M reads per sample being used in publications). In my lab we aim for 40M SE50bp reads. Here an even bigger 10-fold difference in read depth makes a massive difference to the final cost. RNA-seq is one of the applications I’m most interested in and we’ve done array comparisons that show 20M reads is fine for DGE, and fewer can be better if you’ve got lots (>4) of replicates.
- Amplicons: (£262, answers averaged out to £50-450). We use Fluidigm’s Access Arrays but there are dozens of amplicon-seq methods
Sequencing vs library prep: 85% of entrants said library prep was the gold bauble and sequencing was red. This was the easiest question to answer from my perspective as for almost every NGS application being run today sequencing costs are significantly higher than library prep.
Methods: Questions were weighted, scored and summed to give a percentage of the “correct” answers as defined in the table. Question 1 accounted for 50% of points, question 2, 10% and question 3, 40%.
No comments:
Post a Comment
Note: only a member of this blog may post a comment.