I was at the 3rd NGS congress in London on Monday and Tuesday this week and one of the topics we discussed in questions was quantitation of Illumina sequencing libraries. It is still a challenge for many labs and results in varying yields. The people speaking thought that between 5-25% of possible yield was being missed through poor quantification.
Illumina recommend a final concentration of 10–13 pM to get optimum cluster density from v3 cluster kits. There is a hug sample prep spike in NGS technologies where a samples is adapter ligated and massively amplified so a robust quantification can allow the correct amount of library to be added to the flowcell or picotitre-plate. If a sensitive enough system is sued then no-PCR libraries can be used. Most people are stil using PCR amplification and lots of the biases have been removed with protocol improvements.
The method of DNA quantitation is important (no-one wants to run titration flow cells). There are many methods that can be used and I thought I 'd give a run down of the pro's and cons' for each of these (see below). The LOQ values are taken from
Digital PCR provides sensitive and absolute calibration for high throughput sequencing, and I have ordered systems by sensitivity (lowest to highest).
Which one to use: Most labs choose the method that suits them best and this is dependant on skills and experience and also what equipment is available for them to use. However even in the best labs getting cluster density spot on has not been perfected and methods could still be improved (I'm currently working on a solution).
In my lab we find that careful use of the Bioanalyser gives us quantitative and qualitative information from just 1 ul of sample. I think we may move to qPCR now we are making all libraries using TruSeq.
Why is this important? If you agree that 5-20% of achievable yield is being missed then we can work out how many Human genomes we could be sequencing with that unused capacity. To work this out I made some assumptions about the kind of runs people are performing and use PE100 as the standard. On GAIIx I used 50Gb as the yield and 12 days as the run time, for HiSeq I used 250Gb and 10 days. There are currently 529 GAIIx and 425 HiSeq instruments worldwide according to the
map. I assumed that these
could be used 80% of the time (allowing for maintenance and instrument failures), even though many are used nowhere near that capacity.
Total achievable yield for the world in PE100 sequencing is a staggering 7.5Pb.
Missing just 5% of that through poor quantification loses us 747Tb or about 3500 Human genomes at 100x coverage.
Missing 20% loses us 1500Tb or about 15000 Human genomes at 100x coverage.
We need to do better!
The quantitation technology review:
Agilent Bioanalyser (and others) (LOQ 25ng): The bioanalyser uses a capillary electrophoresis chip to run a virtual gel. Whilst the sensitivity is not as good as qPCR or other methods a significant advantage is the collection of both quantitative and qualitative data from a single run using 1ul of library. The Bioanalyser has been used for over a decade to check RNA quality before microarray experiments. The qualitative analysis allows poor libraries to be discarded before any sequence data are generated and this has saved thousands of lanes of sequencing from being performed unnecessarily.
Bioanalyser quantitation is affected by over or under loading and the kits commonly used (DNA1000 and High-Sensitivity) have upper and lower ranges for quantitation. If samples are above the marker peaks then quantitation may not be correct. Done well this system provides usable and robust quantification.
Many labs will run Bioanalyser even if they prefer a different quantitative assay for determining loading concentrations. New systems are also available from Caliper, Qiagen, Shimazdu and I recently saw a very interesting instrument from
Advanced Analytical which we are looking at.
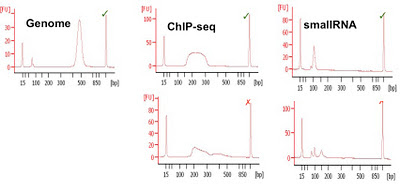 |
| Examples of Bioanalyser libraries (good and bad) from CRI |
UV spectrophotometry (LOQ 2ng): Probably the worst kind of tool to use for sequencing library quantification. Spectrophotometry is affected by contaminants and will report a quantity based on absorbance by anything in the tube. For the purpose of library quantification we are only interested in adapter ligated PCR products, yet primers and other contaminants will skew the results. As a result quantification is almost always inaccurate.
This is the only platform I would recommend you do not use.
Fluorescent detection (LOQ 1ng): Qubit and other plate based fluorometer
use dyes that bind specifically to DNA , ssDNA or RNA and a known standard (take care when making this up) to determine a quantitative estimate of the test samples actual concentration. The Qubit uses Molecular Probes fluorescent dyes which emit signals ONLY when bound to specific target molecules, even at low concentrations. There are some useful resources on the Invitrogen website and a
comparison of Qubit to nanodrop.
I don't think it's nice to bash another technology but the Qubit is simply better for this task.
 |
| Qubit from Invitrogen website |
You can use any plate reading fluorometer and may already have one lurking in your lab or institute.
qPCR (LOQ 0.3-0.003fg): Quantitative PCR (qPCR) is a method of quantifying DNA based on PCR. During a qPCR run intensity data are collected after each PCR cycle from either probes (think TaqMan) or intercalating chemistry (think SYBR). The intensity is directly related to the number of molecules present in the reaction at that cycle and this is a function of the starting amount of DNA. Typically a standard curve (take care when making this up) is run and unknown test samples are compared to the curve to determine a quantitative estimate of the samples actual concentration. qPCR is incredibly sensitive and quite specific and is the method most people recommend.
You can use any qPCR machine and either design your own assay or use a commercial one. You don't need to buy an Illumina qPCR machine or thier kit, just use the onw available in your lab or one next door and spend the money saved on another genome or three!
In the Illumina qPCR quantification protocol they use a polymerase, dNTPs, and two primers designed to the adapter sequences. The primer and adapter sequences are available from Illumina TechSupport but you do have to ask for the and they should not be generally shared (I don't know why they don't just put them on the web, everyone who wants to know does). The design of the assay means that only adapter ligated PCR products should amplify and you will get a very good estimate of concentration fro cluster density. Adapter dimers and other concatamers may also amplify so you need to make sure your sample is not contaminated with too much of these. Illumina also demonstrated that you can use a dissociation curve to determine GC content of your library. You can use this protocol as a starting point for your own if you like.
 |
| Illumina qPCR workflow |
 |
| GC estimation by dissociation curve |
Digital PCR (LOQ 0.03fg): Fluidigm's digital PCR platform has been released for library quanitifaction as the SlingShot kit, available for both Illumina and 454. This kit does not require a calibrator samples and uses positive well counts to determine a quantitative estimate of the samples actual concentration. A sinlge qPCR reaction is setup and loaded onto the Fluidigm chip. This reaction gets partitioned into 765 9nl chambers for PCR. The DNA is loaded at a concentration that results in many wells having no template present. The count of positive wells after PCR is directly related to starting input and quantitation s very sensitive.
 |
| SlingShot image from Fluidigm brochure |
The biggest drawback is the need to buy a very expensive piece of hardware and this technology has only been adopted by labs using Fluidigm for other applications or in some large facilities.
Those are pretty big numbers and many more genomes than are being sequenced in all but the largest of consortia led projects.














{kind=link}
{kind=link}
{kind=link}
{kind=link}