Prof Steve Jackson presented a great seminar today at CRI on his labs work on DNA damage repair. One of the numbers that jumped out at me was 1,000,000, that is 1M DNA damage events per cell per day.
Just think, even with all that going on it still takes most of us 60 years to get cancer! 63% of cancers are diagnosed in people aged 65 or older.
Prof Jackson was also one of the founders of KuDOS Pharmaceuticals Ltd. This venture was acquired by Astra Zeneca in 2005 and resulted in the PARP inhibitor Olaparib. Steve gave us a wonderful journey through the incredibly complex world of DNA repair and it's array of protein complexes and regulatory networks. He finished by by talking about teh therapeutic opportunities and how PARP inhibitors can be used to induce a chemo-biologic "synthetic lethality in Cancer cells, whilst being relatively innocuous to normal cells. One of the things I learned is that KuDOS pharmaceuticals took its name from the Ku
protein, which binds to DNA double-strand breaks and creates a
molecular ladder or scaffold that other proteins involved in NHEJ can
bind to.
PARPhylatics: One of Steve's slides showed a graph of how drug toxicity is affected by dose and genotype. There was a 1250x increase in sensitivity to the PARP inhibitor Olaparib for -/- compared to +/- or +/+ BRCA patients. This kind of sensitivity it did make me wonder if low doses of PARP inhibitors might be a sensible prophylactic treatment for specific groups of patients.
I asked a question after Steve's presentation, "might PARP inhibitors be used in patients with germline DNA repair mutations to prevent or delay cancer development". Steve's response was that this may be possible but there are lots of unanswered questions about this kind of use in patients.
Of course patients with these germline mutations can have very agressive treatment in the form of mastectomy (removal of the breasts) and/or oophorectomy (removal of the ovaries). Both of which can significantly reduce a women’s risk of breast or ovarian cancer. What the additional risks might be of PARPhylactics is going to take some time to work out. I'll certainly be keeping my eye out to see what Steve's new company MISSION Therapeutics Ltd does. There are lots of different players in the DNA damage repair pathways and it sounds like many will turn out to be interesting and druggable targets.
Tuesday, 27 November 2012
Monday, 26 November 2012
Facebook photofit app and Dr Evil's DNA disguise
Many many years ago I started up an Affymetrix microarray core lab. One of the early presentations on the 10k genotyping arrays was an epiphany for me. It was clear from the data that copy number would be easily discernible from the technology. I also thought that 10’s of thousands of genotypes, with the possibility of millions to come would allow strong phenotype:genotype correlations to be made. In fact I went to our TechTransfer department with the idea of developing a photofit chip but the idea was discounted as just too difficult with this new and unproven technology. Things don't stand still in Genomics though.
Biotechnology holds the key: In this month’s Nature Biotechnology Caitlin Smith, Stephen Strauss and Laura DeFrancesco published a very interesting feature article on the use of DNA in forensics. All the ideas from ten years ago came back including one that I have been uncomfortable talking about since it occurred to me (see Dr Evil’s DNA Disguise down the page).
The authors give a very good review of current DNA technology for use in forensics. STR profiles have been used for more than twenty years and recent work showed they could be profiled using NGS, the authors missed this reference and I think it is a key one on the road to NGS tests as it provides a potential stepping stone. (see Short-read,high-throughput sequencing technology for STR genotyping). The next big step is likely to come from NGS-genotyping of 1000’s or more SNPs. The article mentions two interesting uses of SNPs, HIrisPLEX, which uses six genes to determine blue or brown eye colour and hair color; and the Identitas SNP-chip for profiling ethnicity, gender, relatedness, geographic ancestry and other phenotypic traits (although their website is woefully lacking on details).
STRs can work on vanishingly small amounts of DNA but Michael Fischer’s group at the Oxford Systems Biology Institute published a method in Genome Research last year for NGS library prep from 20pg of MouseDNA. It should be possible to amplify or select 1000’s of SNPS from these libraries to run a cheap benchtop sequencer analysis.
It looks to me that the days of STR analysis are numbered.
Facebook photofit app: Eric Lander was on a panel discussion at AGBT a few years ago organised by PacBio. He was taking part in a debate on the public release of genome data and said that his daughter (17 at the time) was happy to splash her life over Facebook and may be much less concerned about sharing her genomic data than her father might. If we can buy a 23andMe test for as little as $99, or a genome for $1000 is there a market for a Facebook app that sends you updates on research findings?
And if people put this kind of data if Facebook with its powerful feature recognition tools could we create a photofit app? This is not as crazy as it sounded to the people sat on the same table as me at that dinner! With millions of possible customers and links to friends and family already in the system, FaceBook seems like the next big step for 23andME or DNAncestry and others like them. It won’t take the FBI long to realise how useful the data are. Although the US has somewhat tighter control on this kind of data than the UK, and I thought we were not yet a police state!
Dr Evil’s DNA disguise: This is the idea I’ve had floating around for more than a decade. I’ve been nervous about writing or discussing it as the idea is such a simple one I’d worried that someone might actually use it and there could be some comeback on me. Fortunately the authors of the Nature Biotechnology article have out the idea out there so here I go...
It’s quite simple really. PCR amplify an STR profile from someone unrelated to you and dilute it into a spray bottle. A quick squirt as you are burgling a house and tracking you down as a suspect in a case becomes more difficult. The DNA disguise will massively out compete the small amount of DNA in your fingerprint in the forensic test.
And if you are concerned that SNPs might take over as the default forensic test then why not make a Nextera library. It only cost $60 to make and again PCR amplification will drown out any signal from your own DNA. A side-box in the article mentions work by Nucleix that demonstrates that you can distinguish native DNA by looking at methylation so the disguise may not be perfect.
I will now not only smell nicer during my next seminar, but will also be almost undetectable in a crime scene with my new DNAftershave.
PS: DNAftershave is now available on eBay (with profits going to charity).
PPS: If you drank a PCR reaction would it be detectable as circulating DNA or in urine? Or would it simply be degraded in your stomach?
Friday, 16 November 2012
Tycho Brae, a genome opportunity lost?
Tycho Brae was recently exhumed for the second time. The astronomer died suddenly in 1601 and analysis of hair from a 1901 exhumation suggested he may have been poisoned. The recent exhumation by the University of Southern Denmark disproved this theory, however what exactly did cause his death still remains a mystery.
Why does Core Genomics care about this?
In 2009 I presented at an ABRF pre-meeting workshop on next-generation sequencing. This was the early days and I talked about Illumina while other speakers spoke about 454 and SOLiD. At the end of my talk I briefly mentioned the 1000 genomes project and put up this slide, suggesting that we might be able to sequence the genomes of famous scientists.
 Recently Svante Pääbo's lab at the Max Planck Institute for Evolutionary Anthropology have developed a new library prep method that works on single-stranded DNA where ssDNA is dephosphorylated, denatured, and ligated to a biotinylated adaptors; this is captured on
streptavidin beads for strand-copying with DNA polymerase and a second adaptor
is blunt-end
ligated producing a sequencing ready library.
Recently Svante Pääbo's lab at the Max Planck Institute for Evolutionary Anthropology have developed a new library prep method that works on single-stranded DNA where ssDNA is dephosphorylated, denatured, and ligated to a biotinylated adaptors; this is captured on
streptavidin beads for strand-copying with DNA polymerase and a second adaptor
is blunt-end
ligated producing a sequencing ready library.
I don't know if they kept and samples for DNA analysis but if they did I'd be happy to sequence them.
Can we compile a list of celebrity scientists (deceased)? Feel free to add names by commenting on this post.
Why does Core Genomics care about this?
In 2009 I presented at an ABRF pre-meeting workshop on next-generation sequencing. This was the early days and I talked about Illumina while other speakers spoke about 454 and SOLiD. At the end of my talk I briefly mentioned the 1000 genomes project and put up this slide, suggesting that we might be able to sequence the genomes of famous scientists.

I don't know if they kept and samples for DNA analysis but if they did I'd be happy to sequence them.
Can we compile a list of celebrity scientists (deceased)? Feel free to add names by commenting on this post.
Wednesday, 14 November 2012
MiSeq: possible growth potential part 4
Our MiSeq has just been replaced after some performance issues and we are now getting ready to start running the upgraded version with the newest chemistry. Hopefully we'll be getting the 10Gb+ that Illumina reported some users are achieving.
In a GenomeWeb article yesterday Illumina responded to Life Technologies recent Proton updates (P1 80M, P2 4580M P3 1.2B sensors). MiSeq read lengths are increasing to 2x300bp and the number of reads will jump from 15-17M to 25M or more making 22Gb possible.
Way back in the Summer of 2011 I speculated that MiSeq would get to 25Gb by imaging more area and generating more reads (see MiSeq 1,), I have continued to follow the systems developments (see here, here and here) and am watching to see what else we might expect in the future. The MiSeq was sold as a box with a lot of potential locked down for easy upgrades and an 'Applesque' development path. Much of what has been made possible recently was probably available from launch, or at least considered by the designers.
Currently only one of the two "lanes" in the MiSeq is being imaged (imagine the return path as a separate lane). Further increases could come from bigger lanes or an upgrade to movable camera stage (unless there is one inside the box already). All pretty simple.
But how far does Illumina need to push MiSeq? With the 2500 rapid runs allowing the same degree of flexibility (on instrument clustering and 24 hour run times), any further increases to MiSeq make the GA dead-in-the-water. Perhaps Illumina will be driven by announcements by Life Technologies? Could Miseq ever deliver really fast run times like PGM or Proton? I'd say it does that already, a single-end 120bp run is completed in under 2 hours if you ignore clustering. Combined with Nextera genome prep you can get sample to genome in a working day.
So what should Illumina focus on next: My bet is that users will want the longest reads possible and we will learn to do new things with Sanger read lengths but tens of millions of them. Any de novo sequencing will be made much easier, structural variation is simplified and splicing becomes as trivial as differential gene expression by read counting (almost). And all of these could possibly be done with lower quality reads. Imagine a 100bp read with Q40 in the first and last 250bp, Q30 from 250-450 & 550-750 and Q10-20 in the middle?
In a GenomeWeb article yesterday Illumina responded to Life Technologies recent Proton updates (P1 80M, P2 4580M P3 1.2B sensors). MiSeq read lengths are increasing to 2x300bp and the number of reads will jump from 15-17M to 25M or more making 22Gb possible.
Way back in the Summer of 2011 I speculated that MiSeq would get to 25Gb by imaging more area and generating more reads (see MiSeq 1,), I have continued to follow the systems developments (see here, here and here) and am watching to see what else we might expect in the future. The MiSeq was sold as a box with a lot of potential locked down for easy upgrades and an 'Applesque' development path. Much of what has been made possible recently was probably available from launch, or at least considered by the designers.
Currently only one of the two "lanes" in the MiSeq is being imaged (imagine the return path as a separate lane). Further increases could come from bigger lanes or an upgrade to movable camera stage (unless there is one inside the box already). All pretty simple.
But how far does Illumina need to push MiSeq? With the 2500 rapid runs allowing the same degree of flexibility (on instrument clustering and 24 hour run times), any further increases to MiSeq make the GA dead-in-the-water. Perhaps Illumina will be driven by announcements by Life Technologies? Could Miseq ever deliver really fast run times like PGM or Proton? I'd say it does that already, a single-end 120bp run is completed in under 2 hours if you ignore clustering. Combined with Nextera genome prep you can get sample to genome in a working day.
So what should Illumina focus on next: My bet is that users will want the longest reads possible and we will learn to do new things with Sanger read lengths but tens of millions of them. Any de novo sequencing will be made much easier, structural variation is simplified and splicing becomes as trivial as differential gene expression by read counting (almost). And all of these could possibly be done with lower quality reads. Imagine a 100bp read with Q40 in the first and last 250bp, Q30 from 250-450 & 550-750 and Q10-20 in the middle?
Wednesday, 7 November 2012
Cancer profiling: NGS is not the only tool in the box
Many groups are working on next-generation sequencing tests for mutation profiling of tumours. There are two main approaches, amplicon sequencing (reviewed in a previous post) and in-solution capture capture (reviewed here and here); both of which only look at DNA which is not immediately limiting, and tests are being commercialised (see my recent Foundation Medicines post).
Of course DNA and NGS is not the only assay that can be used. Other tests target more than this, e.g. FISH for DNA rearrangements, RT-PCR for gene expression or IHC for protein expression.
There are companies aiming to be as comprehensive as possible and one of them also has an interactive tool I thought you may not be aware of.
Cancer pathways: The Caris website has a nice interactive tool to look at Breast, Lung and Colon pathways and the drugs that impact treatment.


Caris Life Sciences have two main technologies, Target NOW and Carisome microvesicle technologies. They have also just selected the MiSeq and TruSeq Custom Amplicon for their sequence based analysis of FFPE tissue. In Illumina's press release Matt Posard said that Caris had completed over 40,000 molecular profiling tests.
Target Now is a molecular profiling
service for personalised cancer medicine. It is based on markers
identified in the literature which correlate with drug response and also
identifies open clinical trials patients may be enrolled in. It is
somewhat similar to the approach used by foundation Medicine and also by
Illumina in their clinical sequencing tests. As well as identifying
patient treatment options Target Now aims to suggest alternative
therapies for patients where the
standard of care is not an option, has been exhausted, or for cases of
highly aggressive,
metastatic, or rare tumours. You can see what is included in their Target Now Breast Cancer test here. It offers a mix of Sequencing, IHC, FISH/CISH, RT-PCR and RFLP. You can also download a sample report from their website.
Carisome technology is a blood based assay for
circulating microvesicles (cMV) or exosomes that can act as cancer
biomarkers. cMVs are nm-sized sub-cellular membrane-bound vesicles
released by many cell types into almost all intracellular fluids,
including blood. These can be proteomically profiled to determine
cell-of-origin. Caris Life Sciences are commercialising cMV profiling as
a potential tool for cancer diagnosis.
Their Executive Director Dr. Daniel Von Hoff (also physician in chief at TGen and Professor of medicine at Mayo) presented at this years ASCO. You can see his presentation on YouTube.
Saturday, 3 November 2012
Session 4: Functional genomics of cancer
Go back to the summary of the CRI symposium.
Carlos Caldas, Functional genomics of cancer: perturbation experiments in the lab and in the clinic. Carlos is a senior group leader at the Cancer Research UK Cambridge Research Institute where I work, he is also a practising oncologist and one of the founders of CRI. Carlos stood in at short notice for Eric Lander who had to stay behind to advise Barack Obama, during the upcoming election!
Carlos Caldas, Functional genomics of cancer: perturbation experiments in the lab and in the clinic. Carlos is a senior group leader at the Cancer Research UK Cambridge Research Institute where I work, he is also a practising oncologist and one of the founders of CRI. Carlos stood in at short notice for Eric Lander who had to stay behind to advise Barack Obama, during the upcoming election!
Carlos started by asking the audience where we can get a definition of functional genomics? Wikipedia! Carlos started with a brief overview of the work the speakers in the session have been instrumental in pushing forward. he spent some time talking about Gary Nolan's work in single cell mass-cytometry.
 |
| Mass-cytometry image from Bendall et al: Science 2011 |
When thinking about functional genomics Carlos thinks about perturbation experiments and presents a good case for the fact that we are already doing this in the clinic, in clinical trials and in xenograft experiments. There are multiple genomes in any cancer and evolution of subclones is driven by therapy as much as normal development.
Carlos showed an analysis of TCGA Breast cancer data looking for the ten subtypes presented in his groups 2012 Nature paper. In the TCGA data the subtypes stand out in a similar manner. Also presented work from Alex Bruna in his lab showing that the effects of perturbations are cell-autonomous context dependant. Clearly cell context is very important when performing a perturbation. Alex Bruna has also been performing xenograft experiments from metastatic breast cancer, trying to co-register what can be seen in mouse xenografts and in patients.
Analysis of clonal evolution in triple negative breast cancer suggests that heterogeneity is not necessarily spatial so DNA extraction from solid tumours can capture a lot of the intra-tumour heterogeneity. Using targeted sequencing we can use cancer exomes in plasma to understand the mechanisms of resistance. Understanding these mechanisms is absolutely key to deciding which combinations of therapy are likely to be most effective in treating patients.
René Bernards, Finding effective combination therapies for cancer through functional genetic. Prof Bernards is Head of Molecular Carcinogenesis at the The Netherlands Cancer Institute.
He p.resented work on differential response of BRAF inhibitionin BRAF mutant melanoma versus colon cancer, the same drug and same genotype in colon cancer has unfavourable response. Why? The group used a synthetic-lethal screen of all kinases by RNAi. Infect cells with shRNA library, culture in presence or absence of drug, look for PCR-amplified barcodes that tag shRNAs for deep-sequencing. EGFR turned out to be the highest likelihood target but is upstream of BRAF, this creates a conundrum.
There is a synergistic response of BRAF V600E colon cancer cells. Vemurafenib leads to increased p-EGFR and other changes in expression. Adding EGFR inhibitors as well lead to a combination therapy that hopefully work well in a clinical setting. Pre-clinical work in mouse showed a clear response in xenograft tumours. When the group looked closely at larger numbers of colon cancer cell lines they saw two which responded to mono-therapy whilst all others were resistant. these two both were negative for EGFR and as such more similar to melanoma cells in their response.
This could suggest that melanoma patients might escape BRAF inhibitor treatment by upregulating EGFR. They showed that this does happen in histopathological analysis of BRAF inhibitor resistant patients and have started a clinical trial to address this.
Dale Porter, Using genomic data coupled with preclinical response data to guide clinical development of IAP antagonists. Dr Porter is Head of ? at Novartis.
IAP antagnoisists, inhibitors of apoptosis by binding to caspases, SMAC can repress this inhibition by binding to the IAP proteins. At Novartis they have been searching for XIAP agonists LCL161 is teh fruit of their search. There are 7 or8 bona-fide IAP family members, ideally we would knock out all of them. Wehn testing LCL161 it became clear that although designed to one IAP they actually inactivate another family member at much higher efficiencies, and that this family member does not bind caspases! The drug appears to work by inducing TNF driven apoptosis. They are aiming to kill cancer cells rather than just stop growth.
Only 7% of cell lines are sensitive to LCL161, is it possible to predict which cell lines or even patients will respond? They are using data and cell lines generated for the cancer cell line encyclopedia and presented a gene expression signature for response to LCL161. It is not jst gene expression being run, they are now collecting CNV, methylation, exome-seq etc across 1000 cell lines treated with multiple parameters.
 |
| cancer cell line encyclopedia cell lines |
Cell line encyclopedia was used to look for compounds that might target triple negative breast cancers. Three candidates were found and all were IAP inhibitors. Digging in the data using pooled shRNA analysis of 250 cell lines using representation sequencing after selection shows a novel functional genomics screen that should help in target discovery. They asked if any shRNAs were depleted in triple negative breast cancers vs other breast cancer cell lines? Again XIAP targeting shRNA came up as targets.
Rebecca McIntyre, High-throughput functional validation of candidate colorectal cancer genes. Rebecca is a PhD student in David Adams research group at the Wellcome Trust Sanger Institute.
Rebecca presented work on validation of three colorectal cancer genes. Colon crypts are very well studied and complex organs. Hyperactivation of WNT pathway is often seen in CRC. The 1990's brought us the canonical APC, Kras, Smad4, TP53 Fearon and Vogelstein model. More recent work in Science 2006 & 2007 identified novel genes and four fo these were selected for validation in APCmin mouse models.
MLL3: a chromatin modifier which was the fourth most mutated CRC gene, it is a large gene with multiple polyA regions both of these make it more likely to be mutated. Is it a bona-fide CRC driver gene? ChIP-seq analysis shows that MLL3 down regulates AXIN2 expression which is involved in WNT signalling. Crypts of MLL3 deficient mice are longer and may also explain why tumouregensis is higher. Rebecca also presented work on PKDH1 and PARK2.
Jason Carroll, Understanding oestrogen receptor transcription in breast cancer. Jason is a senior group leader at the Cancer Research UK Cambridge Research Institute where I work.
Key questions in the Carroll lab:
Nearly all work in ER is being performed in MCF7 and of course this can not possibly represent all ER biology. Work in Jason's lab also uses primary breast cancer tumours and ER ChIP-seq showed clear classification of samples as good, poor or metastatic. Differential binding was seen in good vs poor vs metastatic samples. For three tumours they split the sample in two to get some idea of the impact of tumour heterogeneity, replicates gave highly concordant data. They looked to see if the differential binding switched on different genes but did not have RNA for analysis. They developed a gene predictor and tested it in multiple cohorts where it was shown to correlate with outcome, but only in ER+ve samples.
ERE and Forkhead motifs turn up time and again in poor outcome samples. Data from three different ER+ve cell lines show that generally when FOXA1 moves in the genome ER binding follows. Differential binding also occurs in tamoxifen resistant cell lines when compared to susceptible
ER binding can be reprogrammed in a rapid timescale. Growth factors EGF and IGF can induce tamixofen resistance in just five minutes by reprogramming ER binding in the genome. But what happens to FOXA1? It turns out that mitogen induced ER binding correlates with FOXA1, so what allows FOXA1 to bind?
How do you find the pioneer factors pioneer factor? Using a modified Chromatin IP and proteomic analysis of the total bound protein. They specified that three replicates had to show the same results and that IG controls must be silent. They found 108 ER protein:protein interactions and can do this in patient samples. For FOXA1 they found 24 proteins, many of which were chromatin modifiers and hormone independent preparing FOXA1 for potential ER binding. Are there post-translational modifications of FOXA1? Are there differences in a resistance context?
How important is FOXA1 for ER function? It is essential, if FOXA1 is silenced ER signal drops by 90% even though the protein is still present. Naked DNA oligos will still bind ER as it is not chromatinised. A word of caution for reporter assay interpretation. FOXA1 seems to be the sole determinant of ER function and is one of the signature genes of ER+ve breast cancer (HNF3alpha aka FOXA1). They are now collaborating with Carlos on larger numbers of patient samples.
The big question is whether FOXA1 is going to be a suitable drug target?
"Unanswered questions" panel discussion:
We have the technology to ask big questions about functional genomics of cancer and bring this to the clinic. So how are we going to do this when the scientists do the research, the clinicians have the patients and the drug companies have the drugs?
How will sequencing impact our choices of drug in the clinic?
Can we determine up front which combinations to give at the start of treatment?
Can we better understand which patients should not be treated?
- Where does ER bind to DNA
- How does it bind to DNA
- Are there similarities or differences in ER binding in a resistant or sensitive context?
Nearly all work in ER is being performed in MCF7 and of course this can not possibly represent all ER biology. Work in Jason's lab also uses primary breast cancer tumours and ER ChIP-seq showed clear classification of samples as good, poor or metastatic. Differential binding was seen in good vs poor vs metastatic samples. For three tumours they split the sample in two to get some idea of the impact of tumour heterogeneity, replicates gave highly concordant data. They looked to see if the differential binding switched on different genes but did not have RNA for analysis. They developed a gene predictor and tested it in multiple cohorts where it was shown to correlate with outcome, but only in ER+ve samples.
ERE and Forkhead motifs turn up time and again in poor outcome samples. Data from three different ER+ve cell lines show that generally when FOXA1 moves in the genome ER binding follows. Differential binding also occurs in tamoxifen resistant cell lines when compared to susceptible
ER binding can be reprogrammed in a rapid timescale. Growth factors EGF and IGF can induce tamixofen resistance in just five minutes by reprogramming ER binding in the genome. But what happens to FOXA1? It turns out that mitogen induced ER binding correlates with FOXA1, so what allows FOXA1 to bind?
How do you find the pioneer factors pioneer factor? Using a modified Chromatin IP and proteomic analysis of the total bound protein. They specified that three replicates had to show the same results and that IG controls must be silent. They found 108 ER protein:protein interactions and can do this in patient samples. For FOXA1 they found 24 proteins, many of which were chromatin modifiers and hormone independent preparing FOXA1 for potential ER binding. Are there post-translational modifications of FOXA1? Are there differences in a resistance context?
How important is FOXA1 for ER function? It is essential, if FOXA1 is silenced ER signal drops by 90% even though the protein is still present. Naked DNA oligos will still bind ER as it is not chromatinised. A word of caution for reporter assay interpretation. FOXA1 seems to be the sole determinant of ER function and is one of the signature genes of ER+ve breast cancer (HNF3alpha aka FOXA1). They are now collaborating with Carlos on larger numbers of patient samples.
The big question is whether FOXA1 is going to be a suitable drug target?
"Unanswered questions" panel discussion:
We have the technology to ask big questions about functional genomics of cancer and bring this to the clinic. So how are we going to do this when the scientists do the research, the clinicians have the patients and the drug companies have the drugs?
How will sequencing impact our choices of drug in the clinic?
Can we determine up front which combinations to give at the start of treatment?
Can we better understand which patients should not be treated?
Session 3: The impact of cancer sequencing on medicine
Go back to the summary of the CRI symposium.
Barbara Wold, The genomics discovery path from TCGA and TARGET to the clinic...and back. Unfortunately Dr Wold flew all the way over, escaping hurricane Sandy but was kept in her hotel room by flu.
Keith Peters, was Regius Professor of Physic at the University of Cambridge from 1987 to 2005, where he was also head of the School of Clinical Medicine. Under his leadership the University's Clinical School became a major centre for medical research, complementing Cambridge's strengths in basic biomedical science.
Sir Peters last-minute presentation started with a little look back to 11th April 1988 and a photograph of a document he had written with Sydney Brenner for a meeting with Prime Minister Margaret Thatcher. Sydney Brenner and Keith Peters were "selling" the UK involvement in the Human Genome Project as at the time the MRC was not particularly interested. there were lots of scribbles from Keith and Sydney debating on what the impact would be of the final project on medicine.
Barbara Wold, The genomics discovery path from TCGA and TARGET to the clinic...and back. Unfortunately Dr Wold flew all the way over, escaping hurricane Sandy but was kept in her hotel room by flu.
Keith Peters, was Regius Professor of Physic at the University of Cambridge from 1987 to 2005, where he was also head of the School of Clinical Medicine. Under his leadership the University's Clinical School became a major centre for medical research, complementing Cambridge's strengths in basic biomedical science.
Sir Peters last-minute presentation started with a little look back to 11th April 1988 and a photograph of a document he had written with Sydney Brenner for a meeting with Prime Minister Margaret Thatcher. Sydney Brenner and Keith Peters were "selling" the UK involvement in the Human Genome Project as at the time the MRC was not particularly interested. there were lots of scribbles from Keith and Sydney debating on what the impact would be of the final project on medicine.
Lori Friedman, Overcoming resistance to targeted therapies in breast cancer. A former post-doc with Bruce Ponder and Mary Claire-King now working at Genentech where she is senior director of translational oncology.
Dr Frieman presented two themes in her talk, how mutations can affect choice of therapy, 2nd mechanisms of resistance, innate and acquired and how evolution impact this. Questions; the issue of over-treatment, heterogeneity, what to do with infrequent mutations, druggability of loss-of-function mutations.
She used PI3Kinase as an example; it is the most frequently altered pathway in cancer and PIK3CA is the most commonly mutated druggable oncogene where mutations were identified in 2004 (Samuel's et al Science 2004) and have been shown to increase kinase activity and activate downstream signalling. There are over 20 PIK3CA inhibitors in over 150 clinical trials, a massive investment in a single druggable target!
GDC-0941 is the Genentech PIK3CA inhibitor in clinical trials. Using differential gene expression to determine which samples might be responsive to the drug, 17 gene signature. Suggestive of a feedback mechanism where cells may reset their sensitivity to activation of pathways and become resistant. Clinical protocol starts low and goes higher as tumour cells adapt. When cells get to 10x EC50 single cell clones are selected for analysis to determine mechanisms of resistance. Sequence PIK3CA, GX, CNV, RPPA? and genome sequence. Combination therapy with dual PI3K/mTOR inhibitors rescues resistance to the GDC treatment. Analysis of clones shows PTEN loss and resultant increase of pAKT.
 |
| Cell pathway and mechanistic effects of GDC-0980 treatment in tumor cell lines |
Peter Lichter, Sequencing of paediatric brain tumours: From molecular profiles to clinical translation. Division of Molecular Genetics, at the German Cancer Research Centre.
Pediatric Brain tumours are the most common cause of mortality in children. Dr Lichter spoke about three different paediatric brain tumours and sthe work ongoing at DKFZ as part of the ICGC. Medulloblastoma stratification currently based on age, metastases and degree or resectability, recent genomic works shows consensus (Taylor et al 2012 Acta Neuropathalogia) of four subgroups. Dr Lichter's group wanted to extend the detail of this work. So far they have shown a background mutation rate that is quite low, only around 10 non-synonymous coding mutations and 588 of 765 SNVs were private to an individual (Jones & Jager 2012 Nature). Another word cloud with just a few big words, the cloud was very nicely coloured by subgroup (how did they do that?). There is a correlation between the age of the patient and the number of mutations, not seen in other tumours, what is the mechanism behind this? Their data is suggestive of different subgroups having different cell-of-origin, also global hypomethylation patterns show subgroup specific profiles.
 |
| Peter Lichter's word cloud coloured by stratification group |
Marlous Hoogstraat, moving towards implementation of next-gen into clinical decision making. PhD student at UMC Utrecht, centre for personalised cancer treatment and the research school Cancer Genomics & Developmental Biology.
Marlous presented a collaboration between three centres NKI, UMC Utrecht and Erasmus Medical Center. She described their sequencing approach where they are allocating patients with metastatic disease into clinical trials, taking 2-4 matched biopsies, performing pathological analysis, DNA isolation and then completing a patient stratification using Amplicon sequencing on Ion Torrent and MiSeq. They try to allocate patients to a trial and monitor resistance or progression, taking repeated biopsies to understand results. All in two weeks! They are running 5500 and starting up wildfire to sequence 2000 genes and are moving to whole exome.
The rate limiting step for the analysis is not the sequencing. The logistics are a hurdle, collection and transport of samples, pathological review, batching for processing and sequencing all add time, whilst the sequencing itself is completed in a day or two.
Biopsy protocol: perform image guided biopsy, pathologist selects tumour rich areas for DNA extraction. All tissue is snap frozen. DNA yield and tumour percentage across the first 100 samples showed generally more than 1.5ug of DNA and most tumours are 50% cancer cells although they are using samples with just 20% tumour in very high depth targeted sequencing. Presented results from Sunitinib trial, TP53, KRAS, APC, KDR, Met, CTNNB1, FLT3, PTEN, EGFR, FGFR3, SMAD4, IDH1. They found different numbers of mutations per patient and 11 that have no mutations (silent genomic signature?), there were no strong associations with aggressive or indolent disease. Now moving to Stratification 2.0, including complete tumour suppressors and amplifications.
Data being reported back after multi-disciplinary team meeting (how much do these meetings cost?) The 2000 genes being sequenced in their research track analysis are being captured using a custom Agilent SureSelect product.
Nitzan Rosenfeld, Circulating tumour DNA as a non-invasive tool for cancer diagnostics and research. Nitzan is a group leader at the Cancer Research UK Cambridge Research Institute where I work.
He presented his groups work on ctDNA mutational analysis. Cancer is an evolving disease of the genome and ctDNA provides a way to monitor this in real-time. A problem is the ctDNA contains normal DNA potentially in a high background. His talk started with an experiment where a tumour was initially sequenced for personalised genomic biomarkers and then ctDNA was analysed for these markers in a follow-up study. Comparing ctDNA to CA125 as a marker for high-grade serous ovarian cancer and demonstrating that ctDNA can be informative when CA125 fails. The patients were analysed with digital PCR over 600 and 300 days during which they were treated with secondary and tertiary therapies as tumours relapsed. ctDNA gave a faster sharper response with greater significance in a prognostic setting. The response is probably even sharper but the study is limited by the sampling of blood being 3 weeks after therapy.
His group is also collaborating with Carlos Caldas and Sarah-Jane Dawson. And he presented results that suggest in metastatic breast cancer patients rising ctDNA levels often predate radiological disease progression or a change in CTCs or CA15-3 levels.
He also spoke about a third collaboration showing how it is also possible to look for mutations that could be used in targeted therapy. The collaboration with Tan Min Chen Singapore, looks at acquired resistance to anti-EGFR therapies.
Targeted amplicon sequencing TAm-seq is a practical compromise that allows a sizeable and useful portion of the genome to be sequenced with similar sensitivity to single-locus assays. And it is dirt cheap! How much can we get from ctDNA, only time wil tell?
Bruce Ponder, How can we use knowledge of genetic variation? Bruce is Director of the Cancer Research UK Cambridge Research Institute where I work.
Inherited cancer predisposition is becoming more and more a computational problem but it is important to understand the clinical challenges as well. Cancer susceptibility is well understood in a familial BRCA1/2 context, but only counts for about 15-20% of risk and under 5% of Breast cancers. Missing heritability is buried in the normal genetic variation. In 2000 there were 12 predisposition loci, today over 1000 in 165 different diseases. Consider the predisposition genotypes as a hand of cards dealt at birth, your hand will put you somewhere on the normal distribution. Understanding the mechanism of action of the individual loci will help us better understand cancer, ideally we can understand risk as well and target therapy better.
FGFR2 per allele risk is low at just 1.26% of GWAS heritability, what does this mean in the clinic? Normal lifetime Breast cancer risk is 9.7%, a single copy of the FGFR2 variant increases this to 10.2% and two copies is still just 13.4%. This is not huge.
Breast cancer screening as an example of how genetic screening can affect current methods (see Population based screening in the era of genomics). NHS screens women at age 47, genetic reclassification may have clinical and economic benefit.
Introducing “attributable fraction”: Prof Ponder gave the example of Breast cancer where the attributable fraction of being a women is 99%, so if we got rid of women we would reduce the number of breast cancers massively. Obviously this is not a practical solution. FGFR2 has a significant attributable risk and if reduced to normal would eliminate 16% of breast cancer. Great for public health but what are the effects of taking FGFR2 inhibitors over a long term? But there are good approaches in other diseases, e.g. statins that target the activity of a single enzyme to lower cholesterol and reduce heart disease.
"Unanswered questions" panel discussion introduced by Kieth Peters
"Unanswered questions" panel discussion introduced by Kieth Peters
Is prediction of individual risk by SNPs currently clinically useful?
How will we do clinical trials on what are quickly becoming orphan diseases due to stratification?
How can we target tumour suppressors or other undruggable targets? Is synthetic lethality.
How do you get the dose right in a combination therapy?
How do you generate a combination therapy when your competition has the combinatorial?
Is drug development "self-harming" in that drug companies are all
chasing the same target where we will ultimately need combination
therapies to reduce the rate of resistance so those companies should work harder on different targets and collaborate on combinations?
Session 2: Challenges facing the computational biology of cancer sequencing
Go back to the summary of the CRI symposium.
Overall summary: Is it possible to move the goal-posts and say that a clinical “test” no longer revolves around the actual sequencing of a cancer gene, as in the case of BRCA1/2 screening, but is now more about the algorithm used to ask specific questions of what could be a lifetime stored data set?
How do we better find mutations of very low prevalence? It is possible to see incredibly low prevalence in NGS data but only if we know where to look.
Ewan Birney, ENCODE: Understanding our genome. Dr Birney is Associate Director of the EMBL-European Bioinformatics Institute (EMBL-EBI). He is one of the founders of the Ensembl genome browser and played a key founding and leadership role in the ENCODE project.
Dr Birney gave an overview of the recent ENCODE findings, 182 cell lines/tissues, 164 assays (114 ChIPs), 3010 experiments, 5TB, 1716x coverage of a human genome! A vast amount of data. ENCODE used a uniform analysis pipeline and shows that nearly the whole genome is active if you look at enough cell lines and factors, 98% is within 8kb of an active site.
Replication of experiments is a hot topic in almost every lab, usually because we want to use a few replicates as possible to keep costs down. Unfortunately samples variability gets in the way and more replicates is usually better. However replicates are variable and show discordance and statisticians get absorbed in variance analysis and can ask for more than is practical. For ENCODE Peter Bickel’s team developed consistency analysis (more commonly referred to as IDR analysis), which uses just two replicates. IDR poses that if two replicates are measuring the same biology then genuine signals should be consistent while noise should not be so. This consistency provides a marker between signal and noise and allows you to rank significance.
Discovering functional genome segments: He presented analysis using unstructured ENCODE data to look to see what falls out of the analysis. Promoters were obvious, enhancers and insulators were also found. New discoveries were specific gene end motifs, probably having something to do with poly-adenylation. Confirmation analysis of enhancers was about 50% in both mouse and fish analysis. Ewan took a little diversion in his talk to introduce is to the usefulness of doughnuts as a useful mathematical surface for self organising maps and work Ali Mortazavi has done using SOMs to analyse Gene Ontology terms.
Data is available to share and visualise in both UCSC and Ensembl. The audience were encouraged to fly European!
Jan Korbel, Mechanisms of genomic structural variation in germline and cancer. Dr Korbel is a group leader at EMBL Heidelberg. His lab is both experimental and computational and is focused on understanding heritable genomic structural variants (SVs) in cancer.
Structural variations are the main focus of Dr Korbel's research and he developed the paired-end mapping approaches, read depth, split reads, etc. Applied these tools as part of the 1000 genomes project. The NGS data allow us to see base-pair resolution and understand the functional relevance of these variants, overlay of gene expression data with 1000 genomes data allowed identification of the causative variant for over 1000 eQTLs.
Structural variation can be categorised into four main categories; Non-homologous recombination NAHR, Mobile element insertion (MEI), variable number of simple tandem repeats (VNTR) non-homologous rearrangement. And structural variation hotspots in the human genome are formed by single processes at each hotspot, that is the underlying sequence drives the kind of SV seen in a hotspot.
Some structural variants are difficult to analyse, e.g. balanced polymorphic inversions with no copy number change. Looked for inversion calls in 1000 genomes data and saw? PCR validations showed that all three methods used for analysis had inadequacies. They developed a mixed analysis of read-pair and split-read mapping to identify truly polymorphic inversions and distinguish true from false inversions.
In the second part of his talk Dr Korbel presented his groups contribution to the ICGC PedBrain tumour project. The first tumours they sequenced had mutational phenotypes that went against a gradual model of tumour evolution, no mutations on some chromosomes but massively instable (chromothriptic) chromosome 15. This patient had Li-Fraumeni syndrome and TP53 mutation (Cell 2012 Rausch and see image below). When they looked at the levels of copy number aberration in [patient sot call chromothripsis they found 5/6 cases which were undiagnosed Li-Fraumein syndrome. They were able to show that chromothripsis leads to oncogene amplification presumably by the formation of new circular mini-chromosome. Medulloblastoma development frequently involves one step catastrophic alterations (chromothripsis), linked with TP53 mutations. And results in the formation of double minute minichromosomes.
What are the biological mechanisms behind chromothripsis? Analysis in the Korbel lab suggests that it is a chromosomal shattering that is occurring. Careful monitoring and tumour screening of these patients leads to increased survival rates, and personalised treatments as chemotherapy and/or radiotherapy in the context of germline TP53 mutation could generate secondary tumours through therapy.
How do they do their sequencing? The Korbel group use a mixed sequencing approach using paired-end and mate-pair library prep. This is the methods section from their 2012 Cell paper on Pediatric Medulloblastoma. "DNA library preparation was carried out using Illumina, Inc., paired-end (PE) and mate-pair (MP, or long-range paired-end mapping) protocols. In brief, 5ug (PE) or 10ug (MP) of genomic DNA isolation's were fragmented to ∼300 bp (PE) insert-size with a Covaris device, or to ∼4kb (MP) with a Hydroshear device, followed by size selection through agarose gel excision. Deep sequencing was carried out with Genome Analyzer IIx and HiSeq2000 instruments. Exome capturing was carried out with Agilent SureSelect Human All Exon 50 Mb in-solution capture reagents (vendor's protocol v2.0.1)."
Elaine Mardis, Genomic heterogeneity in cancer cells. Dr. Mardis is director of technology development at the Genome Institute, WashU. Her lab has helped create many of the methods and automation pipelines we use for sequencing the human genome.
Dr Mardis presented the workflow of WGS, this was the first talk to do so and it always helps to make sure the basics are understood. She also reminded us how much the technology has moved on in the last five years. Her lab has developed methods for custom hybrid capture to sequence variant sites at 1000x coverage, this works out very cost effective compare to PCR when you have 1000’s of variants to verify. Older mutations are present at higher variant allele frequency (VAF) so we can model tumour heterogeneity and aim to determine the subclone mutational profile. VAF changes over time or therapy. She showed some very nice visualisations of heterogeneity estimation.
She went on to describe a study of metastatic evolution in Breast cancer, where multiple metastatic lesions were removed post-mortem. Circos plots showed different structural rearrangements at each site and building an evolutionary tree shows the relatedness and distance of the different samples.
Lastly Dr Mardis went on to present the effect of Aromoatse inhibitors on tumour evolution. The reason for the study was to determine a genomic profile indicative of AI response or resistance. The study took Ki67 analysis by biopsy neo-adjuvant and also at surgery, 22 patients had whole genome sequence from the tumour and the initial biopsy core. Comparison of pre and post- treatment genomes showed tumours can have sub-clones emerging or disappearing with AI treatment in responsive patients. However in resistant patients, we still need to find druggable targets as the standard of care is not working for these patients.
HyunChul Jung, Systematic survey of cancer-associated somatic SNPs. He is working on raising the awareness of the dangers faced with most common filtering approaches to find significant SNPs.
Elliott Margulies, Delivering clinically relevant genome information. Dr Margulies is a director of scientific research at Illumina and before that was head of the Genome Informatics at NHGRI.
Illumina wants to bring an end-to-end workflow for clinical sequencing from samples to clinical reports. In his talk he presented some of the technological innovations and how they are being used in the clinic.
Sample prep improvements: New methods coming from Illumina in the New Year will allow us to sequence gel and PCR-free from as little as 100ng of genomic DNA, in just 4.5 hours. Data is of at least he same quality if not even higher than standard TruSeq. Library diversity is significantly improved. The techniques apply well to FFPE DNA as well. Gaps in genome coverage from standard preps appear to be better, particularly in GC rich regions.
Sequencing improvements: The big change coming into labs in the next coupe of months is the HiSeq 2500 (see previous blogs). This will allow the genome-in-a-day, or 27 hours to be precise. And coming alongside this is the new iSAAC alignment algorithm that can align a whole geonome in about 3-5 hours with a sorted deduplicated indel realigned BAM file and a new variant caller called Starling, which will analyse the BAM file in about 3hr to produce a gVCF. All on a $5000 unix box! This analysis will be rolled out into BaseSpace for cloud informatics. Comparison to other methods was done using CEPH trio sequencing and looking for Mendelian conflicts, perhaps the best way to asses the quality of analysis. The new pipeline does as well as GATK but in 3 rather than 36 hours! InDel calling improvement is now the top priority for further developments. On top of this Illumina are pulling in data from multiple public databases, dbSNP, COSMIC, ensemble, UCSC, ENCODE, 1000genomes, genome.gov, Hgmd, etc and generate the annotated gVCF file of 1Gb.
What does it mean for sequencing: All this allows a genome sequence and analysis in 48 hours. Most of us are taking longer to make a single TruSeq library in that time!
But what about analysis and interpretation: Illumina are also focusing on interpretation of the data. Ultimately if we want to make use of this in the clinic it has to be actionable in some way or other. Moving the goal-post to say that the “test” no longer revolves around the actual sequencing but is more about the algorithm used to ask specific questions of what could be a lifetime stored data set. Dr Margulies presented the case of a 5 year old child presented with phenotypic disease, whole genome sequencing and analysis got to six variants in 2 days and a result suggesting Menkes syndrome that was confirmed by the clinic. The data came from Childrens Mercy Hospital, see Saunders et al.
He also presented surveillance of CLL using WGS and targeted sequencing over time (collaboration with Dr Anna Schuh, Oxford). Amplicons were sequenced on MiSeq with TSCA, it is possible to see incredibly low prevalence mutations but only if we know where to look.
Dr Margulies finished with a vision for how we may be sequenced during our lives starting with WGS at birth and with followup analysis and resequencing over time and during disease progression and treatment. Sequencing may be performed in different ways at different stages.
What is a gVCF file? It is a way to encode the entire genome, not just variants. Concatenate regions of similar depth and quality ntp a single record and report minimum values, a genome compression.
Richard Durbin, Human genetic variation: from population sequencing to cellular function. Dr Durbin is joint head of Human Genetics at the Sanger Institute and leads the Genome Informatics group. He has been instrumental in the 1000 Genomes project.
Dr Durbin gave an update on the 1000 genome project and presented work on the genetics of cellular function (miRNA QTLs and CTCF ChIP-seq QTLs). 1092 genomes sequenced at 4x WGS and deep exome-seq. Over 2500, from over 25 populations have been collected for continued sequencing. The project so far has generated a wealth of data, currently about 20Tb of data. One of the computational challenges is the visualisation of variation in complex datasets.
"Unanswered questions" panel discussion introduced by Ewan Birney
Can 1000genomes help us develop better algorithms for InDel detection?
What are the panels views on the challenges that need to be addressed to integrate the clinical interpretation with analysis?
Overall summary: Is it possible to move the goal-posts and say that a clinical “test” no longer revolves around the actual sequencing of a cancer gene, as in the case of BRCA1/2 screening, but is now more about the algorithm used to ask specific questions of what could be a lifetime stored data set?
How do we better find mutations of very low prevalence? It is possible to see incredibly low prevalence in NGS data but only if we know where to look.
Ewan Birney, ENCODE: Understanding our genome. Dr Birney is Associate Director of the EMBL-European Bioinformatics Institute (EMBL-EBI). He is one of the founders of the Ensembl genome browser and played a key founding and leadership role in the ENCODE project.
Dr Birney gave an overview of the recent ENCODE findings, 182 cell lines/tissues, 164 assays (114 ChIPs), 3010 experiments, 5TB, 1716x coverage of a human genome! A vast amount of data. ENCODE used a uniform analysis pipeline and shows that nearly the whole genome is active if you look at enough cell lines and factors, 98% is within 8kb of an active site.
Replication of experiments is a hot topic in almost every lab, usually because we want to use a few replicates as possible to keep costs down. Unfortunately samples variability gets in the way and more replicates is usually better. However replicates are variable and show discordance and statisticians get absorbed in variance analysis and can ask for more than is practical. For ENCODE Peter Bickel’s team developed consistency analysis (more commonly referred to as IDR analysis), which uses just two replicates. IDR poses that if two replicates are measuring the same biology then genuine signals should be consistent while noise should not be so. This consistency provides a marker between signal and noise and allows you to rank significance.
Discovering functional genome segments: He presented analysis using unstructured ENCODE data to look to see what falls out of the analysis. Promoters were obvious, enhancers and insulators were also found. New discoveries were specific gene end motifs, probably having something to do with poly-adenylation. Confirmation analysis of enhancers was about 50% in both mouse and fish analysis. Ewan took a little diversion in his talk to introduce is to the usefulness of doughnuts as a useful mathematical surface for self organising maps and work Ali Mortazavi has done using SOMs to analyse Gene Ontology terms.
Data is available to share and visualise in both UCSC and Ensembl. The audience were encouraged to fly European!
Jan Korbel, Mechanisms of genomic structural variation in germline and cancer. Dr Korbel is a group leader at EMBL Heidelberg. His lab is both experimental and computational and is focused on understanding heritable genomic structural variants (SVs) in cancer.
Structural variations are the main focus of Dr Korbel's research and he developed the paired-end mapping approaches, read depth, split reads, etc. Applied these tools as part of the 1000 genomes project. The NGS data allow us to see base-pair resolution and understand the functional relevance of these variants, overlay of gene expression data with 1000 genomes data allowed identification of the causative variant for over 1000 eQTLs.
Structural variation can be categorised into four main categories; Non-homologous recombination NAHR, Mobile element insertion (MEI), variable number of simple tandem repeats (VNTR) non-homologous rearrangement. And structural variation hotspots in the human genome are formed by single processes at each hotspot, that is the underlying sequence drives the kind of SV seen in a hotspot.
Some structural variants are difficult to analyse, e.g. balanced polymorphic inversions with no copy number change. Looked for inversion calls in 1000 genomes data and saw? PCR validations showed that all three methods used for analysis had inadequacies. They developed a mixed analysis of read-pair and split-read mapping to identify truly polymorphic inversions and distinguish true from false inversions.
In the second part of his talk Dr Korbel presented his groups contribution to the ICGC PedBrain tumour project. The first tumours they sequenced had mutational phenotypes that went against a gradual model of tumour evolution, no mutations on some chromosomes but massively instable (chromothriptic) chromosome 15. This patient had Li-Fraumeni syndrome and TP53 mutation (Cell 2012 Rausch and see image below). When they looked at the levels of copy number aberration in [patient sot call chromothripsis they found 5/6 cases which were undiagnosed Li-Fraumein syndrome. They were able to show that chromothripsis leads to oncogene amplification presumably by the formation of new circular mini-chromosome. Medulloblastoma development frequently involves one step catastrophic alterations (chromothripsis), linked with TP53 mutations. And results in the formation of double minute minichromosomes.
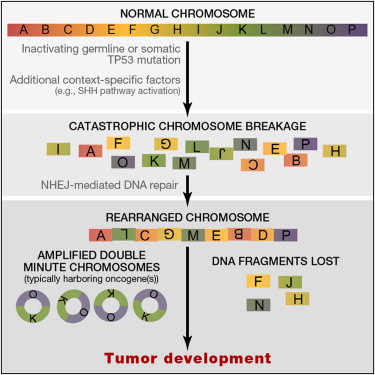 |
| Rausch 2012 Graphical abstract |
What are the biological mechanisms behind chromothripsis? Analysis in the Korbel lab suggests that it is a chromosomal shattering that is occurring. Careful monitoring and tumour screening of these patients leads to increased survival rates, and personalised treatments as chemotherapy and/or radiotherapy in the context of germline TP53 mutation could generate secondary tumours through therapy.
How do they do their sequencing? The Korbel group use a mixed sequencing approach using paired-end and mate-pair library prep. This is the methods section from their 2012 Cell paper on Pediatric Medulloblastoma. "DNA library preparation was carried out using Illumina, Inc., paired-end (PE) and mate-pair (MP, or long-range paired-end mapping) protocols. In brief, 5ug (PE) or 10ug (MP) of genomic DNA isolation's were fragmented to ∼300 bp (PE) insert-size with a Covaris device, or to ∼4kb (MP) with a Hydroshear device, followed by size selection through agarose gel excision. Deep sequencing was carried out with Genome Analyzer IIx and HiSeq2000 instruments. Exome capturing was carried out with Agilent SureSelect Human All Exon 50 Mb in-solution capture reagents (vendor's protocol v2.0.1)."
Elaine Mardis, Genomic heterogeneity in cancer cells. Dr. Mardis is director of technology development at the Genome Institute, WashU. Her lab has helped create many of the methods and automation pipelines we use for sequencing the human genome.
Dr Mardis presented the workflow of WGS, this was the first talk to do so and it always helps to make sure the basics are understood. She also reminded us how much the technology has moved on in the last five years. Her lab has developed methods for custom hybrid capture to sequence variant sites at 1000x coverage, this works out very cost effective compare to PCR when you have 1000’s of variants to verify. Older mutations are present at higher variant allele frequency (VAF) so we can model tumour heterogeneity and aim to determine the subclone mutational profile. VAF changes over time or therapy. She showed some very nice visualisations of heterogeneity estimation.
She went on to describe a study of metastatic evolution in Breast cancer, where multiple metastatic lesions were removed post-mortem. Circos plots showed different structural rearrangements at each site and building an evolutionary tree shows the relatedness and distance of the different samples.
Lastly Dr Mardis went on to present the effect of Aromoatse inhibitors on tumour evolution. The reason for the study was to determine a genomic profile indicative of AI response or resistance. The study took Ki67 analysis by biopsy neo-adjuvant and also at surgery, 22 patients had whole genome sequence from the tumour and the initial biopsy core. Comparison of pre and post- treatment genomes showed tumours can have sub-clones emerging or disappearing with AI treatment in responsive patients. However in resistant patients, we still need to find druggable targets as the standard of care is not working for these patients.
HyunChul Jung, Systematic survey of cancer-associated somatic SNPs. He is working on raising the awareness of the dangers faced with most common filtering approaches to find significant SNPs.
Elliott Margulies, Delivering clinically relevant genome information. Dr Margulies is a director of scientific research at Illumina and before that was head of the Genome Informatics at NHGRI.
Illumina wants to bring an end-to-end workflow for clinical sequencing from samples to clinical reports. In his talk he presented some of the technological innovations and how they are being used in the clinic.
Sample prep improvements: New methods coming from Illumina in the New Year will allow us to sequence gel and PCR-free from as little as 100ng of genomic DNA, in just 4.5 hours. Data is of at least he same quality if not even higher than standard TruSeq. Library diversity is significantly improved. The techniques apply well to FFPE DNA as well. Gaps in genome coverage from standard preps appear to be better, particularly in GC rich regions.
Sequencing improvements: The big change coming into labs in the next coupe of months is the HiSeq 2500 (see previous blogs). This will allow the genome-in-a-day, or 27 hours to be precise. And coming alongside this is the new iSAAC alignment algorithm that can align a whole geonome in about 3-5 hours with a sorted deduplicated indel realigned BAM file and a new variant caller called Starling, which will analyse the BAM file in about 3hr to produce a gVCF. All on a $5000 unix box! This analysis will be rolled out into BaseSpace for cloud informatics. Comparison to other methods was done using CEPH trio sequencing and looking for Mendelian conflicts, perhaps the best way to asses the quality of analysis. The new pipeline does as well as GATK but in 3 rather than 36 hours! InDel calling improvement is now the top priority for further developments. On top of this Illumina are pulling in data from multiple public databases, dbSNP, COSMIC, ensemble, UCSC, ENCODE, 1000genomes, genome.gov, Hgmd, etc and generate the annotated gVCF file of 1Gb.
What does it mean for sequencing: All this allows a genome sequence and analysis in 48 hours. Most of us are taking longer to make a single TruSeq library in that time!
But what about analysis and interpretation: Illumina are also focusing on interpretation of the data. Ultimately if we want to make use of this in the clinic it has to be actionable in some way or other. Moving the goal-post to say that the “test” no longer revolves around the actual sequencing but is more about the algorithm used to ask specific questions of what could be a lifetime stored data set. Dr Margulies presented the case of a 5 year old child presented with phenotypic disease, whole genome sequencing and analysis got to six variants in 2 days and a result suggesting Menkes syndrome that was confirmed by the clinic. The data came from Childrens Mercy Hospital, see Saunders et al.
He also presented surveillance of CLL using WGS and targeted sequencing over time (collaboration with Dr Anna Schuh, Oxford). Amplicons were sequenced on MiSeq with TSCA, it is possible to see incredibly low prevalence mutations but only if we know where to look.
Dr Margulies finished with a vision for how we may be sequenced during our lives starting with WGS at birth and with followup analysis and resequencing over time and during disease progression and treatment. Sequencing may be performed in different ways at different stages.
What is a gVCF file? It is a way to encode the entire genome, not just variants. Concatenate regions of similar depth and quality ntp a single record and report minimum values, a genome compression.
Richard Durbin, Human genetic variation: from population sequencing to cellular function. Dr Durbin is joint head of Human Genetics at the Sanger Institute and leads the Genome Informatics group. He has been instrumental in the 1000 Genomes project.
Dr Durbin gave an update on the 1000 genome project and presented work on the genetics of cellular function (miRNA QTLs and CTCF ChIP-seq QTLs). 1092 genomes sequenced at 4x WGS and deep exome-seq. Over 2500, from over 25 populations have been collected for continued sequencing. The project so far has generated a wealth of data, currently about 20Tb of data. One of the computational challenges is the visualisation of variation in complex datasets.
"Unanswered questions" panel discussion introduced by Ewan Birney
Can 1000genomes help us develop better algorithms for InDel detection?
What are the panels views on the challenges that need to be addressed to integrate the clinical interpretation with analysis?
Session 1: Cancer genome sequencing
Go back to the summary of the CRI symposium.
Overall summary: The top10 BrCA CaGens only account for 50% of mutation load, the tail is going to be important in personalising treatment. Some patients only show a single driver mutation, can cancer be driven by a single gene?
Overall summary: The top10 BrCA CaGens only account for 50% of mutation load, the tail is going to be important in personalising treatment. Some patients only show a single driver mutation, can cancer be driven by a single gene?
Can the cancer genome as a molecular fossil record of the tumour?
A common image in many of the presentations is the tailed distribution of cancer drivers. Can the shape of these tails tell us anything, do different cancers give the same shaped tail? And why do we call them tails anyway? The term comes from an image drawn by the statistician William Sealy Gosset to demonstrate different distributions from the mean. Cancer drivers appear to be leptokurtic, from the Greek leptos, meaning 'thin' versus the fatter tailed platykurtic distribution. His sketches of a platypus and two kangaroos are the origin of the term tail.
 |
| Platykurtic and Leptokurtic tails |
Welcome from Bruce Ponder: This is the 5th annual CRI symposium and previous symposia have been successful partly due to the format where 45 minutes are reserved for panel discussion at the end of each session. Not your usual didactic question and answer session, we want to focus on the things we don’t know so please don’t be shy to ask a question where you don’t know the answer!
Eric lander needed to stay in Washington due to the election as he is a presidential advisor. Brad Bernstein had to stay as a tree squashed his house!
Mike Stratton, The evolution of the cancer genome: Shankar Balasubramanian introduced Prfo Stratton by saying he was ”one of the early birds of Cancer genome sequencing”. Prof Stratton is director Sanger Institute, joint head of the Cancer Genome Project, and Professor of Cancer Genetics at the Institute of Cancer Research. His lab has made major advances in understanding the genetics of cancer including the identification of BRCA2 and the first whole Cancer genome sequencing papers on Lung and Melanoma.
Become common thought that emergence of cancer is a Darwinian evolution, from fertilised egg to cancer cell it is possible to determine a lineage of cancer evolution. During the whole lineage many somatic mutagenic exposures lead to mutator phenotype and full-blown cancer. We now classify mutations as Drivers and Passengers, 5-7 drivers suggested but recent data give us a better picture still perhaps no more than 10. Drivers sit in a sea of passengers 10’s to 10000’s that do not confer a clonal-expansion advantage. Sequencing reveals everything.
Sequencing is the primary tool in our understanding of Cancer genomics. Most cancer genes so far discovered are dominant with about 10% recessive mutations.
Prof Stratton presented the landscape of driver mutations in a BrCA exome and CNV analysis of 100 BrCA (80% ER+ / 20% ER-). 9 new CaGenes, most are truncating and inactivating mutations, most of these new genes were recessive genes. Chromatin modifying genes keep coming up in Cancer sequencing studies as the major new class of CaGenes. TP53 and PIK3CA come out top of the list, again. BrCA has 60 or more CaGenes operative but not all contribute to cancer to the same extent, a long tail of infrequently mutated CaGenes. The top10 BrCA CaGens only account for 50% of mutation load, the tail is going to be important in personalising treatment. A greater number of patients that only showed a single driver mutation, somewhat surprising but lots of reasons why others could have been missed. Can cancer be driven by a single gene?
In his presentation he discussed three ER+ve patients with very different genomic landscapes that are likely to have an impact on treatment. There are two schools of thought, 1st only a few pathways being abrogated so disease is still the same in all patients, 2nd each patient is unique and their disease needs to be treated as such. The next few years should allow us to say which statement is closer to the truth for major cancers.
He showed exome results of all substitutions, this is not a normal distribution some patients have 8 mutations others have almost 800. The same was seen in 21 BrCa WGS with between 3k to 180k somatic mutations, why such a difference? He also showed big differences in the kind of base substitutions found, something is different in these patients, but what does it mean for treatment of BrCa if anything? They looked at triplet frequencies of 5’ and 3’ bases in a mutational context. For 3’ base substitutions it is clear that CpG dinucleotide mutations are important in many patients. However 5’ bases can be important in TpC context whch is something we are not so familiar with. This kind of analysis is teasing out new mutational processes that are gong to require lots of investigations. The team cnied the term kataegis (thunderstom) for a pattern of localised hypermutation and hypothesised that the APOBEC gene family could be responsible for this mutational phenotype.
 |
| Kataegis |
New analysis found five major mutational processes; signatures A-E using algorithms designed to detect features in faces. BRCA1 & 2 BrCa’s appear to be D & E driven mutational phenotypes with little CpG substitution. It is also possible to see that the mutational profile changes over time of cancer development. Signature A is operative early but less contributory in later cancer.
Localised mutational signatures: most mutations are shown to be distributed across the genome, but some kataegic cancers produce Manhattan plot style profiles where mutations appear to cluser to localised regions of the genome. Most have a TpC context and also co-cluster with genomic rearrangements. Is the APOBEC repair process responsible for this?
 |
| Rainfall plots, hundreds and thousands anyone? |
The team hypothesise that the APOBEC family of cytidine deaminases (AID, APOBEC3A-H), normally involved in DNA editing are over activated in kataegic cancers. And Prof Stratton clearly presented a putative mechanism of kataegic rearrangement. Why do members of this family target specific regions of the genome. Do they target specific regions or are they secondary and targeting regions where rearrangement has already begun but rapidly increase the damage done? Experiments in yeast where gene are introduced allow discovery of foci of kataegis but there is still a lot to discover in the mechanisms underlying this.
Sean Grimmond, Cohort and personalised cancer genome studies of pancreatic cancer: Prof Grimmond is director of the Queensland Centre for Medical Genomics (QCMG) which opened in 2010. The centres NGS facility is generating pancreatic and ovarian cancer data for ICGC.
Prof Grimmond's title slide was a big word cloud for PaCa, KRAS was big bit there was lots of other stuff in there going on. His lab is taking a cohort based approach to try and find drivers and mechanisms of PaCa. Pancreas Lung, Oesophagus, Brain, Stomach, Myeloma have all shown very small improvements in life expectancy over the last 40 years, we need to understand these cancers better to help patients.
Big cohorts allow us to find out lots about Cancer but at some level Cancer is a personal disease. The PaCa analysed at QCMG so far come from only 20% of patients who were eligible for a “Whipple” operation and have a higher 5-year survival. 450 patients consented, 400 samples were collected, 100 xenografts have been created and blood is being collected longitudinally. Patients can opt-in to get back all data and every patient so far has asked for this. I think this demonstrates how strong patient desire for knowledge is.
Xenografts remove the normal contamination that complicates WGS analysis but Xenografts are under massive selection and can be genomically unstable. Primaries are best but we should not discount xenografts. PaCa tumour cellularity can be very poor and median in the analysis presented was just 30%. We need to maximise sensitivity and accuracy of WGS. snpCGH analysis and KRAS analysis of exons 2 & 3 allow good cellularity calling. The team also run mixing experiments to generate a “truth” and see how analytical methods perform.
Seq, seq and more seq: Genomes, exomes, transcriptomes and epigenome (450k arrays). LIMs for the whole process! Data made available as part of ICGC. They discovered 16 PaCa drivers, five of which were new candidates. Will blood based screening for these 16 PaCa drivers be enough to find patients earlier potentially whilst still operable and hopefully increase 5 year survival rates? They also found many recurrent structural variants and are now taking forward 200 genes for custom “exome” approach to sequencing.
ROBO-SLIT: is a new pathway in PaCa shown to be important in other cancers, where it is frequently lost. ROBO 1&2 are receptors for the SLIT ligand and can decrease cell adhesion, and increase wnt activity and cell motility. Expression of ROBO2 can be used to determine survival, ROBO2 low expression has poor outcome, but ROBO3 high expression has the same affect.
How does all this discovery impact patients? Xenografts can be considered as “mouse avatars”. Some patients are outliers for expression of transporters that confer resistance or susceptibility to Gemcitabine, as shown by treatment of the xenograft. One patient showed a positive response and when PaCa relapsed treatment with gemcitabine put them back into remission, now over a year with the disease, quite extraordinary for PaCa. The team are discovering actionable mutations (BRCA2, HER2, etc) but it is difficult to get this data back in time to treat patients with novel drugs. HER2 is amplified in 2-3% of PaCa patients making them candidates for treatment with anti-HER2 therapy Tarceva, unfortunately a patient being treated had therapy stopped and they died just four weeks later. there is a clear need to get data back fast, especially in PaCa. IMPACT is a study for individualised molecular pancreatic cancer therapy, recruiting actionable patients for individualised treatment.
Seishi Ogawa, Genetic analysis of myeloid neoplasms in childhood: University of Tokyo, cancer genomics project.
Compromised blood cell production, predisposition to AML, >10,000 new patients per year in USA, cure only by allogeneic hematopoietic stem cell transplantation. Again driver mutations are important and several candidates are well understood and found in other cancers. Whole exome sequencing of 50 cases and WGS of 6 cases found 421 somatic mutations with 9.2 per sample (quite low when compared to solid tumours). Splicing factors, Epigenetic regulators, RAS pathway and Transcription genes all implicated as new candidates. Splicesome mutations are found in many other cancers at low levels 5-15% with mutations in all components of the splicing machinery, as hotspots or across whole genes.
Used targeted sequencing of 105 candidate genes across 730 patients and all major subtypes to find over 2000 mutations with frequencies of >5%-30%. Graphs of cancer drivers always have a tail, does this tail tell us anything about cancer drivers?
Binay Panda, Landscape of genetic changes in oral tongue squamous carcinoma: Dr Panda works at Ganit Labs, a new genome sequencing and translational genomics lab in Bangalore, India; just two years old.
Head and Nck cancer 6th most important cancer in global incidence, 95% are squamous cell carcinomas. There is a huge disparity in survival rates in India vs rest of the world because most patients present with late stage disease. Dr Panda’s talk focused on oral tongue squamous carcinoma (OTSC). His interest in this particular form of head and neck cancer is due to its increasing incidence when compared to reducing incidence of other H&N cancers. OTSC has a similar developmental timeline to colon cancer, they would like to understand why some patches of disease progress to full-blown OTSC. They are using Exome-seq, WGS & snpCGH in tumour:normal paired analysis, there is some difficulty in selecting a true matched normal tissue so they are usually using blood lymphocytes.
HPV –ve tumours have a higher mutation rate than HPV +ve’s and HPV is implicated in 35% of oral tongue squamous carcinoma. TP53, Notch1 & Jak3 are three of the most highly mutated genes in OTSC for single-nucleotide variation, other genes show fewer SNV’s but CNVs.
The understanding of cancer genomics sequencing and personalised genomics medicine by Indian doctors is low. Bringing these technologies to India is going to require big changes in implementation and cost. Dr Panda suggested we move from a Western Bugatti Veyron to an Indian Tata Nano model, where complete re-engineering and design produces a technological tool that best fits the subcontinent.
Carlos López-Otín, The genomic landscape of chronic lymphocytic leukemia: Dr López-Otín is based in the Universidad de Oviedo and IUOPA, Spain. His primary interest is the proteolytic enzymes involved in human pathology and cancer progression. His work has shown that these enzymes can have both pro- and anti-tumour activity, and that it may be possible to develop therapeutic approaches for human disease targeting proteolytic enzymes.
CLL-ICGC in Spain, pilot project looked at 2 aggressive and 2 indolent CLL patients. Cells were sorted to allow tumour:normal comparison, and the major variants have been followed up in over 300 patients. They saw an enormous diversity in the number of somatic mutations with high and low subtypes separated by the number of mutations in the variable region of IG genes, recurrent NOTCH1, MYD88 and XPO1 mutations are drivers in CLL disease evolution. MYD88 was shown to be an activating mutation in 3% of CLL cases. All Notch1 mutations are found in the same PEST degradation region, leading to hyper regulation and poor prognosis with transformation of the disease to a very aggressive syndrome.
In order to sequence larger numbers of patients the work is moving from WGS to whole exome sequencing and now 105 patients have been completed. Mutated genes are grouped in particular pathways. Dr López-Otín showed another wod cloud but for CLL the image is very much less crowded than Prof Grimond’s PaCa one. He also showed another word cloud form a different paper showing some similarities but also some obvious differences which he put down to stratification of patients sequenced. This underscores the goals of the ICGC to sequence a large enough number of patients to discover the majority of cancer drivers.
His lab is also looking at epigenomic changes using 450k arrays and some whole genome bisulfite sequencing. Unmutated and mutated CLL split into two groups with respect to methylation, this has clinical relevance in patient prognosis due to cell of origin. There are three group of patients separated according to their cancer deriving from naive B cells (NBCs), memory B cells or an intermediary cell type. Hopefully this will lead to new treatments for CLL.
An offshoot of the exome-seq program has looked at rare diseases and discovered a new hereditary progeroid syndrome, Néstor-Guillermo progeria syndrome implicating a new gene BANF1.
Levi Garraway, Models of tumour evolution: biological and therapeutic implications: Dr. Levi A. Garraway is based at the Dana-Farber Cancer Institute and the Broad Institute. His lab focuses on melanoma and prostate cancer looking at the reasons behind those cancers, mechanisms of resistance to therapy and personalising cancer medicine.
Dr Garrawy started by summarising thematically what has been said today. Several speakers updates different malignancies and present catalogues and insights into novel tumour biology, and discover markers and mechanisms of therapeutic response and resistance. He described the cancer genome as a molecular fossil record of the tumour (is this correct, do the fossils remain?)
Dr Garraway’s primary question was is it possible to stop tumours adapting? His melanoma slide showed the same tailed distribution on driver genes. Melanoma has one of the highest mutation rates per Mb of all cancers and it is driven by UV damage. The high numbers of mutations creates issue in analysis with over 87000 mutations in 121 exomes and 500 genes mutated in over 10% of samples!
How many drivers is it possible to have?
Most of the genes rated as significant in Melanoma have low expression and have high numbers of silent mutations, this presents a conundrum or a problem in the analysis methods. In order to refine the methods they group approached the background mutation as not genome-uniform, apparently this is a common assumption in other methods. They used the intronic and off-target sequence in exome data to look at the mutation rate in or outside of exons, the ratio of exonic:intronic mutations (InVEx) allows a much more realistic analysis of significant genes and the numbers of these dropped from 700 to a handful. Follow up of these candidates, Rac1, PPP6C, ARID2 is ongoing.
But what is going on in the Melanomas without BRAF or NRAS mutations? NF1 mutations are enriched in about 1/3rd of these, there is still work to do to understand the other 2/3rds. See the paper for more information.
Is Chromothripsis important in the development of Melanoma? Massive catastrophic chromosomal damage could be an alternative model to the gradualist one accepted today. And he presented a third model of “punctuated” evolution driven by chromosomal chain events as seen in Prostate Cancer? Work by Sylvan Baca an MD-PhD student is developing a novel chain-finding algorithm. Analysis of 57 Prostate cancer genomes shows closed-chains in 84% of patients, 2/3rds have multiple chains, chains show sub-clonality and many chains contain cancer genes. There are three models of chromosomal evolution, and this may matter as they could illuminate new prevention strategies, they may throw up novel synthetic-lethal vulnerabilities (PARP-inhibitor like treatments), can chains be used to separate patients into high and low risk subtypes? All of this points to the fact that whole genome analysis needs to be run as exome-seq would have missed this.
Sequencing of nevi (birthmarks or moles) might allow us to understand if melanocytes have a higher background mutation rate than other cells and if this is the case then the high background mutation rate in Melanoma may be a consequence of normal cellular progress.
"Unanswered questions" panel discussion, introduced by Mike Stratton:
Do we have problems finding cancer drivers? The tails do suggest we might not find all drivers.
Is there a recipe for Cancer genome sequencing? >50% cellularity, 30x normal, 50x tumour for WGS. But this has to be a how long is a piece of string question. It very much depends on cellularity and clonality of the tumour.
Will single-cell sequencing revolutionise cancer genomics?
What are the benefits of whole exome sequencing versus whole genome? Cost vs richness of data, vs power!
Do we have problems finding cancer drivers? The tails do suggest we might not find all drivers.
Is there a recipe for Cancer genome sequencing? >50% cellularity, 30x normal, 50x tumour for WGS. But this has to be a how long is a piece of string question. It very much depends on cellularity and clonality of the tumour.
Will single-cell sequencing revolutionise cancer genomics?
What are the benefits of whole exome sequencing versus whole genome? Cost vs richness of data, vs power!
Subscribe to:
Posts (Atom)