Go back to the summary of the CRI symposium.
Overall summary: The top10 BrCA CaGens only account for 50% of mutation load, the tail is
going to be important in personalising treatment. Some
patients only show a single driver mutation, can cancer be
driven by a single gene?
Can the cancer genome as a molecular fossil record of the tumour?
A common image in many of the presentations is the tailed distribution of cancer drivers. Can the shape of these tails tell us anything, do different cancers give the same shaped tail? And why do we call them tails anyway? The term comes from an image drawn by the statistician
William Sealy Gosset to demonstrate different distributions from the mean. Cancer drivers appear to be leptokurtic, from the Greek leptos, meaning 'thin' versus the fatter tailed platykurtic distribution. His sketches of a platypus and two kangaroos are the origin of the term tail.
 |
| Platykurtic and Leptokurtic tails |
Welcome from Bruce Ponder: This is the 5th annual CRI symposium and previous symposia have been successful partly due to the format where 45 minutes are
reserved for panel discussion at the end of each session. Not your usual didactic question and
answer session, we want to focus on the things we don’t know so please don’t be shy to
ask a question where you don’t know the answer!
Eric lander needed to stay in Washington due to the election as he is a presidential advisor. Brad Bernstein had to stay as a tree squashed his house!
Mike Stratton, The evolution of the cancer genome: Shankar Balasubramanian introduced Prfo Stratton by saying he was ”one of the early birds of Cancer genome sequencing”. Prof Stratton is director Sanger Institute, joint head of the Cancer Genome Project, and Professor of Cancer Genetics at the Institute of Cancer Research. His lab has made major advances in understanding the genetics of cancer including the identification of BRCA2 and the first whole Cancer genome sequencing papers on Lung and Melanoma.
Become common thought that emergence of cancer is a Darwinian evolution, from fertilised egg to cancer cell it is possible to determine a lineage of cancer evolution. During the whole lineage many somatic mutagenic exposures lead to mutator phenotype and full-blown cancer. We now classify mutations as Drivers and Passengers, 5-7 drivers suggested but recent data give us a better picture still perhaps no more than 10. Drivers sit in a sea of passengers 10’s to 10000’s that do not confer a clonal-expansion advantage. Sequencing reveals everything.
Sequencing is the primary tool in our understanding of Cancer genomics. Most cancer genes so far discovered are dominant with about 10% recessive mutations.
Prof Stratton presented the landscape of driver mutations in a BrCA exome and CNV analysis of 100 BrCA (80% ER+ / 20% ER-). 9 new CaGenes, most are truncating and inactivating mutations, most of these new genes were recessive genes. Chromatin modifying genes keep coming up in Cancer sequencing studies as the major new class of CaGenes. TP53 and PIK3CA come out top of the list, again. BrCA has 60 or more CaGenes operative but not all contribute to cancer to the same extent, a long tail of infrequently mutated CaGenes. The top10 BrCA CaGens only account for 50% of mutation load, the tail is going to be important in personalising treatment. A greater number of patients that only showed a single driver mutation, somewhat surprising but lots of reasons why others could have been missed. Can cancer be driven by a single gene?
In his presentation he discussed three ER+ve patients with very different genomic landscapes that are likely to have an impact on treatment. There are two schools of thought, 1st only a few pathways being abrogated so disease is still the same in all patients, 2nd each patient is unique and their disease needs to be treated as such. The next few years should allow us to say which statement is closer to the truth for major cancers.
He showed exome results of all substitutions, this is not a normal distribution some patients have 8 mutations others have almost 800. The same was seen in 21 BrCa WGS with between 3k to 180k somatic mutations, why such a difference? He also showed big differences in the kind of base substitutions found, something is different in these patients, but what does it mean for treatment of BrCa if anything? They looked at triplet frequencies of 5’ and 3’ bases in a mutational context. For 3’ base substitutions it is clear that CpG dinucleotide mutations are important in many patients. However 5’ bases can be important in TpC context whch is something we are not so familiar with. This kind of analysis is teasing out new mutational processes that are gong to require lots of investigations. The team cnied the term
kataegis (thunderstom) for a pattern of localised hypermutation and hypothesised that the APOBEC gene family could be responsible for this mutational phenotype.
 |
| Kataegis |
New analysis found five major mutational processes; signatures A-E using algorithms designed to detect features in faces. BRCA1 & 2 BrCa’s appear to be D & E driven mutational phenotypes with little CpG substitution. It is also possible to see that the mutational profile changes over time of cancer development. Signature A is operative early but less contributory in later cancer.
Localised mutational signatures: most mutations are shown to be distributed across the genome, but some kataegic cancers produce Manhattan plot style profiles where mutations appear to cluser to localised regions of the genome. Most have a TpC context and also co-cluster with genomic rearrangements. Is the APOBEC repair process responsible for this?
 |
| Rainfall plots, hundreds and thousands anyone? |
The team hypothesise that the APOBEC family of cytidine deaminases (AID, APOBEC3A-H), normally involved in DNA editing are over activated in kataegic cancers. And Prof Stratton clearly presented a putative mechanism of kataegic rearrangement. Why do members of this family target specific regions of the genome. Do they target specific regions or are they secondary and targeting regions where rearrangement has already begun but rapidly increase the damage done? Experiments in yeast where gene are introduced allow discovery of foci of kataegis but there is still a lot to discover in the mechanisms underlying this.
Sean Grimmond, Cohort and personalised cancer genome studies of pancreatic cancer: Prof Grimmond is director of the
Queensland Centre for Medical Genomics (QCMG) which opened in 2010. The centres NGS facility is generating pancreatic and ovarian cancer data for ICGC.
Prof Grimmond's title slide was a big word cloud for PaCa, KRAS was big bit there was lots of other stuff in there going on. His lab is taking a cohort based approach to try and find drivers and mechanisms of PaCa. Pancreas Lung, Oesophagus, Brain, Stomach, Myeloma have all shown very small improvements in life expectancy over the last 40 years, we need to understand these cancers better to help patients.
Big cohorts allow us to find out lots about Cancer but at some level Cancer is a personal disease. The PaCa analysed at QCMG so far come from only 20% of patients who were eligible for a “
Whipple” operation and have a higher 5-year survival. 450 patients consented, 400 samples were collected, 100 xenografts have been created and blood is being collected longitudinally. Patients can opt-in to get back all data and every patient so far has asked for this. I think this demonstrates how strong patient desire for knowledge is.
Xenografts remove the normal contamination that complicates WGS analysis but Xenografts are under massive selection and can be genomically unstable. Primaries are best but we should not discount xenografts. PaCa tumour cellularity can be very poor and median in the analysis presented was just 30%. We need to maximise sensitivity and accuracy of WGS. snpCGH analysis and KRAS analysis of exons 2 & 3 allow good cellularity calling. The team also run mixing experiments to generate a “truth” and see how analytical methods perform.
Seq, seq and more seq: Genomes, exomes, transcriptomes and epigenome (450k arrays). LIMs for the whole process! Data made available as part of ICGC. They discovered 16 PaCa drivers, five of which were new candidates. Will blood based screening for these 16 PaCa drivers be enough to find patients earlier potentially whilst still operable and hopefully increase 5 year survival rates? They also found many recurrent structural variants and are now taking forward 200 genes for custom “exome” approach to sequencing.
ROBO-SLIT: is a new pathway in PaCa shown to be important in other cancers, where it is frequently lost. ROBO 1&2 are receptors for the SLIT ligand and can decrease cell adhesion, and increase wnt activity and cell motility. Expression of ROBO2 can be used to determine survival, ROBO2 low expression has poor outcome, but ROBO3 high expression has the same affect.
How does all this discovery impact patients? Xenografts can be considered as “
mouse avatars”. Some patients are outliers for expression of transporters that confer resistance or susceptibility to Gemcitabine, as shown by treatment of the xenograft. One patient showed a positive response and when PaCa relapsed treatment with gemcitabine put them back into remission, now over a year with the disease, quite extraordinary for PaCa. The team are discovering actionable mutations (BRCA2, HER2, etc) but it is difficult to get this data back in time to treat patients with novel drugs. HER2 is amplified in 2-3% of PaCa patients making them candidates for treatment with anti-HER2 therapy Tarceva, unfortunately a patient being treated had therapy stopped and they died just four weeks later. there is a clear need to get data back fast, especially in PaCa.
IMPACT is a study for individualised molecular pancreatic cancer therapy, recruiting actionable patients for individualised treatment.
Seishi Ogawa, Genetic analysis of myeloid neoplasms in childhood: University of Tokyo, cancer genomics project.
Compromised blood cell production, predisposition to AML, >10,000 new patients per year in USA, cure only by
allogeneic hematopoietic stem cell transplantation. Again driver mutations are important and several candidates are well understood and found in other cancers. Whole exome sequencing of 50 cases and WGS of 6 cases found 421 somatic mutations with 9.2 per sample (quite low when compared to solid tumours). Splicing factors, Epigenetic regulators, RAS pathway and Transcription genes all implicated as new candidates. Splicesome mutations are found in many other cancers at low levels 5-15% with mutations in all components of the splicing machinery, as hotspots or across whole genes.
Used targeted sequencing of 105 candidate genes across 730 patients and all major subtypes to find over 2000 mutations with frequencies of >5%-30%. Graphs of cancer drivers always have a tail, does this tail tell us anything about cancer drivers?
Binay Panda, Landscape of genetic changes in oral tongue squamous carcinoma: Dr Panda works at Ganit Labs, a new genome sequencing and translational genomics lab in Bangalore, India; just two years old.
Head and Nck cancer 6th most important cancer in global incidence, 95% are squamous cell carcinomas. There is a huge disparity in survival rates in India vs rest of the world because most patients present with late stage disease. Dr Panda’s talk focused on oral tongue squamous carcinoma (OTSC). His interest in this particular form of head and neck cancer is due to its increasing incidence when compared to reducing incidence of other H&N cancers. OTSC has a similar developmental timeline to colon cancer, they would like to understand why some patches of disease progress to full-blown OTSC. They are using Exome-seq, WGS & snpCGH in tumour:normal paired analysis, there is some difficulty in selecting a true matched normal tissue so they are usually using blood lymphocytes.
The understanding of cancer genomics sequencing and personalised genomics medicine by Indian doctors is low. Bringing these technologies to India is going to require big changes in implementation and cost. Dr Panda suggested we move from a Western Bugatti Veyron to an Indian Tata Nano model, where complete re-engineering and design produces a technological tool that best fits the subcontinent.
Carlos López-Otín, The genomic landscape of chronic lymphocytic leukemia: Dr López-Otín is based in the Universidad de Oviedo and IUOPA, Spain. His primary interest is the proteolytic enzymes involved in human pathology and cancer progression. His work has shown that these enzymes can have both pro- and anti-tumour activity, and that it may be possible to develop therapeutic approaches for human disease targeting proteolytic enzymes.
CLL-ICGC in Spain,
pilot project looked at 2 aggressive and 2 indolent CLL patients. Cells were sorted to allow tumour:normal comparison, and the major variants have been followed up in over 300 patients. They saw an enormous diversity in the number of somatic mutations with high and low subtypes separated by the number of mutations in the variable region of IG genes, recurrent
NOTCH1, MYD88 and
XPO1 mutations are drivers in CLL disease evolution.
MYD88 was shown to be an activating mutation in 3% of CLL cases. All
Notch1 mutations are found in the same PEST degradation region, leading to hyper regulation and poor prognosis with transformation of the disease to a very aggressive syndrome.
In order to sequence larger numbers of patients the work is moving from WGS to whole exome sequencing and now
105 patients have been completed. Mutated genes are grouped in particular pathways. Dr López-Otín showed another wod cloud but for CLL the image is very much less crowded than Prof Grimond’s PaCa one. He also showed another word cloud form a different paper showing some similarities but also some obvious differences which he put down to stratification of patients sequenced. This underscores the goals of the ICGC to sequence a large enough number of patients to discover the majority of cancer drivers.
Levi Garraway, Models of tumour evolution: biological and therapeutic implications: Dr. Levi A. Garraway is based at the
Dana-Farber Cancer Institute and the
Broad Institute. His lab focuses on melanoma and prostate cancer looking at the reasons behind those cancers, mechanisms of resistance to therapy and personalising cancer medicine.
Dr Garrawy started by summarising thematically what has been said today. Several speakers updates different malignancies and present catalogues and insights into novel tumour biology, and discover markers and mechanisms of therapeutic response and resistance. He described the cancer genome as a molecular fossil record of the tumour (is this correct, do the fossils remain?)
Dr Garraway’s primary question was is it possible to stop tumours adapting? His melanoma slide showed the same tailed distribution on driver genes. Melanoma has one of the highest mutation rates per Mb of all cancers and it is driven by UV damage. The high numbers of mutations creates issue in analysis with over 87000 mutations in 121 exomes and 500 genes mutated in over 10% of samples!
How many drivers is it possible to have?
Most of the genes rated as significant in Melanoma have low expression and have high numbers of silent mutations, this presents a conundrum or a problem in the analysis methods. In order to refine the methods they group approached the background mutation as not genome-uniform, apparently this is a common assumption in other methods. They used the intronic and off-target sequence in exome data to look at the mutation rate in or outside of exons, the ratio of exonic:intronic mutations (
InVEx) allows a much more realistic analysis of significant genes and the numbers of these dropped from 700 to a handful. Follow up of these candidates, Rac1, PPP6C, ARID2 is ongoing.
But what is going on in the Melanomas without BRAF or NRAS mutations? NF1 mutations are enriched in about 1/3rd of these, there is still work to do to understand the other 2/3rds.
See the paper for more information.
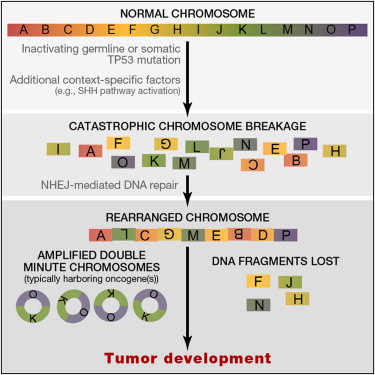
Is Chromothripsis important in the development of Melanoma? Massive catastrophic chromosomal damage could be an alternative model to the gradualist one accepted today. And he presented a third model of “punctuated” evolution driven by chromosomal chain events as seen in Prostate Cancer? Work by Sylvan Baca an MD-PhD student is developing a novel chain-finding algorithm. Analysis of 57 Prostate cancer genomes shows closed-chains in 84% of patients, 2/3rds have multiple chains, chains show sub-clonality and many chains contain cancer genes. There are three models of chromosomal evolution, and this may matter as they could illuminate new prevention strategies, they may throw up novel synthetic-lethal vulnerabilities (PARP-inhibitor like treatments), can chains be used to separate patients into high and low risk subtypes? All of this points to the fact that whole genome analysis needs to be run as exome-seq would have missed this.
Sequencing of
nevi (birthmarks or moles) might allow us to understand if melanocytes have a higher background mutation rate than other cells and if this is the case then the high background mutation rate in Melanoma may be a consequence of normal cellular progress.
"Unanswered questions" panel discussion, introduced by Mike Stratton:
Do we have problems finding cancer drivers? The tails do suggest we might not find all drivers.
Is there a recipe for Cancer genome sequencing? >50% cellularity, 30x normal, 50x tumour for WGS. But this has to be a how long is a piece of string question. It very much depends on cellularity and clonality of the tumour.
Will single-cell sequencing revolutionise cancer genomics?
What are the benefits of whole exome sequencing versus whole genome? Cost vs richness of data, vs power!