Overall summary: Is it possible to move the goal-posts and say that a clinical “test” no longer revolves around the actual sequencing of a cancer gene, as in the case of BRCA1/2 screening, but is now more about the algorithm used to ask specific questions of what could be a lifetime stored data set?
How do we better find mutations of very low prevalence? It is possible to see incredibly low prevalence in NGS data but only if we know where to look.
Ewan Birney, ENCODE: Understanding our genome. Dr Birney is Associate Director of the EMBL-European Bioinformatics Institute (EMBL-EBI). He is one of the founders of the Ensembl genome browser and played a key founding and leadership role in the ENCODE project.
Dr Birney gave an overview of the recent ENCODE findings, 182 cell lines/tissues, 164 assays (114 ChIPs), 3010 experiments, 5TB, 1716x coverage of a human genome! A vast amount of data. ENCODE used a uniform analysis pipeline and shows that nearly the whole genome is active if you look at enough cell lines and factors, 98% is within 8kb of an active site.
Replication of experiments is a hot topic in almost every lab, usually because we want to use a few replicates as possible to keep costs down. Unfortunately samples variability gets in the way and more replicates is usually better. However replicates are variable and show discordance and statisticians get absorbed in variance analysis and can ask for more than is practical. For ENCODE Peter Bickel’s team developed consistency analysis (more commonly referred to as IDR analysis), which uses just two replicates. IDR poses that if two replicates are measuring the same biology then genuine signals should be consistent while noise should not be so. This consistency provides a marker between signal and noise and allows you to rank significance.
Discovering functional genome segments: He presented analysis using unstructured ENCODE data to look to see what falls out of the analysis. Promoters were obvious, enhancers and insulators were also found. New discoveries were specific gene end motifs, probably having something to do with poly-adenylation. Confirmation analysis of enhancers was about 50% in both mouse and fish analysis. Ewan took a little diversion in his talk to introduce is to the usefulness of doughnuts as a useful mathematical surface for self organising maps and work Ali Mortazavi has done using SOMs to analyse Gene Ontology terms.
Data is available to share and visualise in both UCSC and Ensembl. The audience were encouraged to fly European!
Jan Korbel, Mechanisms of genomic structural variation in germline and cancer. Dr Korbel is a group leader at EMBL Heidelberg. His lab is both experimental and computational and is focused on understanding heritable genomic structural variants (SVs) in cancer.
Structural variations are the main focus of Dr Korbel's research and he developed the paired-end mapping approaches, read depth, split reads, etc. Applied these tools as part of the 1000 genomes project. The NGS data allow us to see base-pair resolution and understand the functional relevance of these variants, overlay of gene expression data with 1000 genomes data allowed identification of the causative variant for over 1000 eQTLs.
Structural variation can be categorised into four main categories; Non-homologous recombination NAHR, Mobile element insertion (MEI), variable number of simple tandem repeats (VNTR) non-homologous rearrangement. And structural variation hotspots in the human genome are formed by single processes at each hotspot, that is the underlying sequence drives the kind of SV seen in a hotspot.
Some structural variants are difficult to analyse, e.g. balanced polymorphic inversions with no copy number change. Looked for inversion calls in 1000 genomes data and saw? PCR validations showed that all three methods used for analysis had inadequacies. They developed a mixed analysis of read-pair and split-read mapping to identify truly polymorphic inversions and distinguish true from false inversions.
In the second part of his talk Dr Korbel presented his groups contribution to the ICGC PedBrain tumour project. The first tumours they sequenced had mutational phenotypes that went against a gradual model of tumour evolution, no mutations on some chromosomes but massively instable (chromothriptic) chromosome 15. This patient had Li-Fraumeni syndrome and TP53 mutation (Cell 2012 Rausch and see image below). When they looked at the levels of copy number aberration in [patient sot call chromothripsis they found 5/6 cases which were undiagnosed Li-Fraumein syndrome. They were able to show that chromothripsis leads to oncogene amplification presumably by the formation of new circular mini-chromosome. Medulloblastoma development frequently involves one step catastrophic alterations (chromothripsis), linked with TP53 mutations. And results in the formation of double minute minichromosomes.
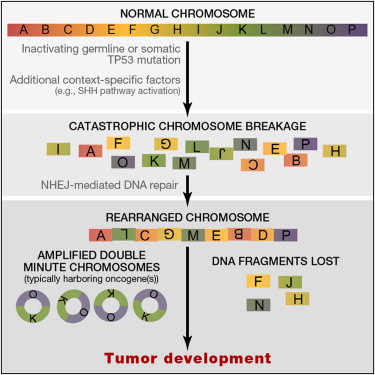 |
| Rausch 2012 Graphical abstract |
What are the biological mechanisms behind chromothripsis? Analysis in the Korbel lab suggests that it is a chromosomal shattering that is occurring. Careful monitoring and tumour screening of these patients leads to increased survival rates, and personalised treatments as chemotherapy and/or radiotherapy in the context of germline TP53 mutation could generate secondary tumours through therapy.
How do they do their sequencing? The Korbel group use a mixed sequencing approach using paired-end and mate-pair library prep. This is the methods section from their 2012 Cell paper on Pediatric Medulloblastoma. "DNA library preparation was carried out using Illumina, Inc., paired-end (PE) and mate-pair (MP, or long-range paired-end mapping) protocols. In brief, 5ug (PE) or 10ug (MP) of genomic DNA isolation's were fragmented to ∼300 bp (PE) insert-size with a Covaris device, or to ∼4kb (MP) with a Hydroshear device, followed by size selection through agarose gel excision. Deep sequencing was carried out with Genome Analyzer IIx and HiSeq2000 instruments. Exome capturing was carried out with Agilent SureSelect Human All Exon 50 Mb in-solution capture reagents (vendor's protocol v2.0.1)."
Elaine Mardis, Genomic heterogeneity in cancer cells. Dr. Mardis is director of technology development at the Genome Institute, WashU. Her lab has helped create many of the methods and automation pipelines we use for sequencing the human genome.
Dr Mardis presented the workflow of WGS, this was the first talk to do so and it always helps to make sure the basics are understood. She also reminded us how much the technology has moved on in the last five years. Her lab has developed methods for custom hybrid capture to sequence variant sites at 1000x coverage, this works out very cost effective compare to PCR when you have 1000’s of variants to verify. Older mutations are present at higher variant allele frequency (VAF) so we can model tumour heterogeneity and aim to determine the subclone mutational profile. VAF changes over time or therapy. She showed some very nice visualisations of heterogeneity estimation.
She went on to describe a study of metastatic evolution in Breast cancer, where multiple metastatic lesions were removed post-mortem. Circos plots showed different structural rearrangements at each site and building an evolutionary tree shows the relatedness and distance of the different samples.
Lastly Dr Mardis went on to present the effect of Aromoatse inhibitors on tumour evolution. The reason for the study was to determine a genomic profile indicative of AI response or resistance. The study took Ki67 analysis by biopsy neo-adjuvant and also at surgery, 22 patients had whole genome sequence from the tumour and the initial biopsy core. Comparison of pre and post- treatment genomes showed tumours can have sub-clones emerging or disappearing with AI treatment in responsive patients. However in resistant patients, we still need to find druggable targets as the standard of care is not working for these patients.
HyunChul Jung, Systematic survey of cancer-associated somatic SNPs. He is working on raising the awareness of the dangers faced with most common filtering approaches to find significant SNPs.
Elliott Margulies, Delivering clinically relevant genome information. Dr Margulies is a director of scientific research at Illumina and before that was head of the Genome Informatics at NHGRI.
Illumina wants to bring an end-to-end workflow for clinical sequencing from samples to clinical reports. In his talk he presented some of the technological innovations and how they are being used in the clinic.
Sample prep improvements: New methods coming from Illumina in the New Year will allow us to sequence gel and PCR-free from as little as 100ng of genomic DNA, in just 4.5 hours. Data is of at least he same quality if not even higher than standard TruSeq. Library diversity is significantly improved. The techniques apply well to FFPE DNA as well. Gaps in genome coverage from standard preps appear to be better, particularly in GC rich regions.
Sequencing improvements: The big change coming into labs in the next coupe of months is the HiSeq 2500 (see previous blogs). This will allow the genome-in-a-day, or 27 hours to be precise. And coming alongside this is the new iSAAC alignment algorithm that can align a whole geonome in about 3-5 hours with a sorted deduplicated indel realigned BAM file and a new variant caller called Starling, which will analyse the BAM file in about 3hr to produce a gVCF. All on a $5000 unix box! This analysis will be rolled out into BaseSpace for cloud informatics. Comparison to other methods was done using CEPH trio sequencing and looking for Mendelian conflicts, perhaps the best way to asses the quality of analysis. The new pipeline does as well as GATK but in 3 rather than 36 hours! InDel calling improvement is now the top priority for further developments. On top of this Illumina are pulling in data from multiple public databases, dbSNP, COSMIC, ensemble, UCSC, ENCODE, 1000genomes, genome.gov, Hgmd, etc and generate the annotated gVCF file of 1Gb.
What does it mean for sequencing: All this allows a genome sequence and analysis in 48 hours. Most of us are taking longer to make a single TruSeq library in that time!
But what about analysis and interpretation: Illumina are also focusing on interpretation of the data. Ultimately if we want to make use of this in the clinic it has to be actionable in some way or other. Moving the goal-post to say that the “test” no longer revolves around the actual sequencing but is more about the algorithm used to ask specific questions of what could be a lifetime stored data set. Dr Margulies presented the case of a 5 year old child presented with phenotypic disease, whole genome sequencing and analysis got to six variants in 2 days and a result suggesting Menkes syndrome that was confirmed by the clinic. The data came from Childrens Mercy Hospital, see Saunders et al.
He also presented surveillance of CLL using WGS and targeted sequencing over time (collaboration with Dr Anna Schuh, Oxford). Amplicons were sequenced on MiSeq with TSCA, it is possible to see incredibly low prevalence mutations but only if we know where to look.
Dr Margulies finished with a vision for how we may be sequenced during our lives starting with WGS at birth and with followup analysis and resequencing over time and during disease progression and treatment. Sequencing may be performed in different ways at different stages.
What is a gVCF file? It is a way to encode the entire genome, not just variants. Concatenate regions of similar depth and quality ntp a single record and report minimum values, a genome compression.
Richard Durbin, Human genetic variation: from population sequencing to cellular function. Dr Durbin is joint head of Human Genetics at the Sanger Institute and leads the Genome Informatics group. He has been instrumental in the 1000 Genomes project.
Dr Durbin gave an update on the 1000 genome project and presented work on the genetics of cellular function (miRNA QTLs and CTCF ChIP-seq QTLs). 1092 genomes sequenced at 4x WGS and deep exome-seq. Over 2500, from over 25 populations have been collected for continued sequencing. The project so far has generated a wealth of data, currently about 20Tb of data. One of the computational challenges is the visualisation of variation in complex datasets.
"Unanswered questions" panel discussion introduced by Ewan Birney
Can 1000genomes help us develop better algorithms for InDel detection?
What are the panels views on the challenges that need to be addressed to integrate the clinical interpretation with analysis?
No comments:
Post a Comment
Note: only a member of this blog may post a comment.